



Chứng loạn dưỡng cơ Duchenne và chứng loạn dưỡng cơ Becker là các bệnh di truyền gen lặn liên kết nhiễm sắc thể X đặc trưng bởi tình trạng yếu cơ đầu gần chi tiến triển do sự thoái hóa của sợi cơ. Chứng loạn dưỡng Becker khởi phát muộn và gây ra các triệu chứng nhẹ hơn. Chẩn đoán được gợi ý bởi lâm sàng và được xác định bằng cách phân tích các sản phẩm protein (loạn dưỡng) của gen đột biến. Điều trị tập trung vào việc duy trì chức năng thông qua vật lý trị liệu và sử dụng nẹp và dụng cụ chỉnh hình. Bệnh nhân mắc chứng loạn dưỡng Duchenne nên được dùng prednisone hoặc deflazacort và đôi khi là liệu pháp di truyền.
Nguồn chủ đề

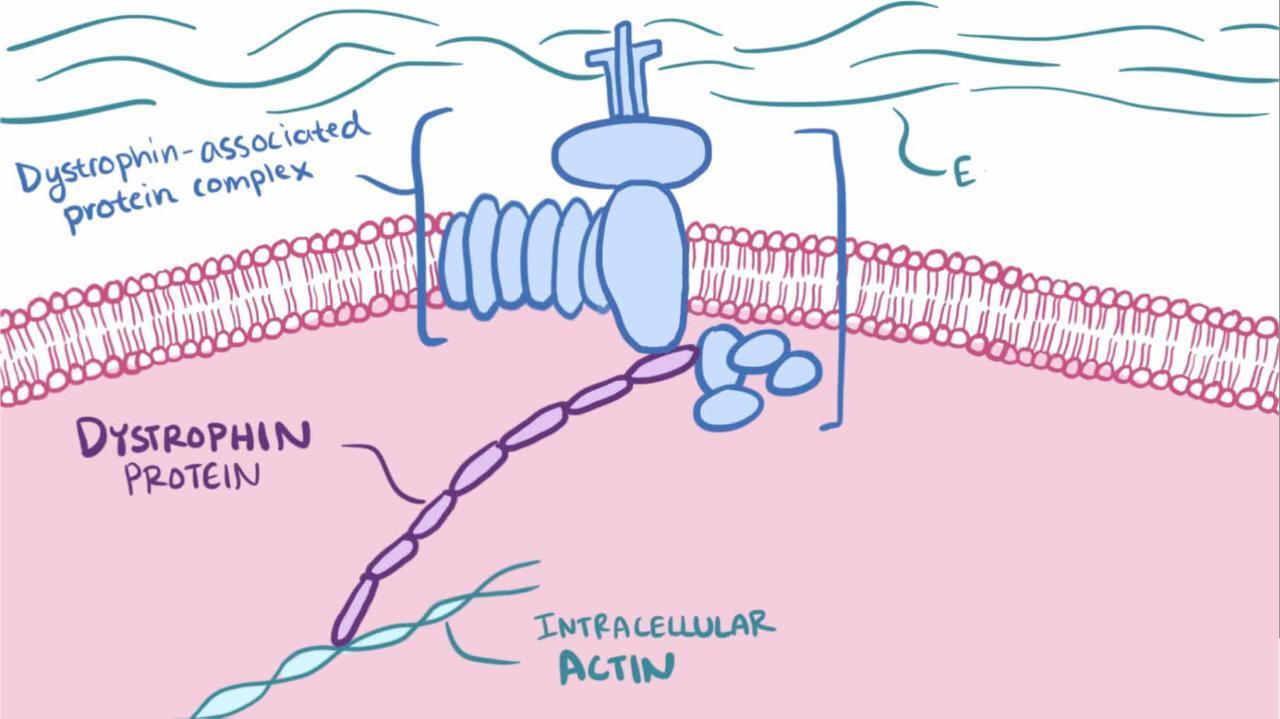
Loạn dưỡng cơ là các rối loạn cơ di truyền, tiến triển do khiếm khuyết của một hoặc nhiều gen cần thiết cho cấu trúc và chức năng cơ bình thường; các thay đổi loạn dưỡng (ví dụ, hoại tử và tái tạo sợi cơ) được nhìn thấy trên các mẫu sinh thiết.
Chứng loạn dưỡng cơ Duchenne và chứng loạn dưỡng cơ Becker là những chứng loạn dưỡng cơ phổ biến nhất. Chúng là do sự đột biến của gen dystrophin, gen người lớn nhất được biết đến, tại locus Xp21.2. Có tới 70% loạn dưỡng Duchenne là do mất một hoặc nhiều exon, khoảng 10% do sao chép và 20% do đột biến điểm. Trong loạn dưỡng Becker, khoảng 70% số bệnh nhân có đột biến mất đoạn, 20% số bệnh nhân có đột biến nhân đôi và có tới 10% số bệnh nhân là đột biến điểm. (1).
Trong chứng loạn dưỡng cơ Duchenne, các đột biến này dẫn đến sự vắng mặt trầm trọng (< 5%) của dystrophin, một protein trong màng tế bào cơ. Trong chứng Becker, các đột biến dẫn đến sự tạo ra dystrophin bất thường hoặc thiếu dystrophin.
Chứng loạn dưỡng Duchenne và chứng loạn dưỡng Becker cùng ảnh hưởng đến khoảng 1/5000 đến 1/6000 ca sinh con trai còn sống (1); phần lớn có Duchenne. Nữ mang gen có thể bị tăng creatine kinase không triệu chứng và có thể phì đại bắp chân.

Tài liệu tham khảo chung
1. Duan D, Goemans N, Takeda S, et al: Duchenne muscular dystrophy. Nat Rev Dis Primers 7(1):13, 2021. doi: 10.1038/s41572-021-00248-3
Triệu chứng và Dấu hiệu
Chứng loạn dưỡng Duchenne
Rối loạn này ảnh hưởng đến khoảng 20/100.000 ca sinh con trai còn sống và biểu hiện điển hình ở độ tuổi từ 2 tuổi đến 3 tuổi. (1). Yếu cơ ảnh hưởng đến cơ gốc chi, thường ở chi dưới đầu tiên. Trẻ em thường biểu hiện đi bằng đầu ngón chân và có dáng đi lắc lư và cột sống lưng quá ưỡn. Họ gặp khó khăn khi chạy, nhảy, leo lên cầu thang, và đứng lên từ sàn nhà. Trẻ em thường xuyên bị ngã, thường gây gãy xương tay hoặc chân (ở khoảng 20% số bệnh nhân) (2). Sự tiến triển của yếu cơ là hằng định, và co cứng các chi và chứng vẹo cột sống gặp ở hầu hết trẻ em. Giả phì đại cơ (sự thay thế chất béo và chất xơ của một số nhóm cơ lớn, đặc biệt là vùng cẳng chân) xuất hiện. Hầu hết trẻ em cần sử dụng xe lăn trước 12 tuổi và nếu không được hỗ trợ thở máy, hầu hết trẻ em sẽ chết vì biến chứng hô hấp ở tuổi 20. Trẻ em được hỗ trợ thở máy có thể sống thêm từ 10 năm đến 20 năm.
Hậu quả của tổn thương đến cơ tim bao gồm bệnh cơ tim giãn, dẫn truyền bất thường, và loạn nhịp tim. Các biến chứng như vậy xuất hiện ở khoảng 1/3 bệnh nhân ở trước tuổi 14 và ở tất cả các bệnh nhân trên 18 tuổi; tuy nhiên, vì những bệnh nhân này không thể tập thể dục, tổn thương của tim thường không có triệu chứng cho đến giai đoạn muộn của bệnh. Khoảng một phần ba có mức độ nhẹ, không tiến triển giảm phát triển trí tuệ, ảnh hưởng đến khả năng lời nói nhiều hơn hoạt động.
Chứng loạn dưỡng cơ Becker
So với chứng loạn dưỡng Duchenne, chứng loạn dưỡng Becker ảnh hưởng đến < 8/100.000 ca sinh là con trai còn sống, thường xuất hiện triệu chứng muộn hơn và nhẹ hơn (3). Vận động thường được bảo tồn cho đến khi ít nhất là 15 tuổi, và nhiều trẻ em vẫn còn vận động được đến tuổi trưởng thành. Hầu hết trẻ em bị ảnh hưởng đều sống đến tuổi 30 và 40.
Tài liệu tham khảo về các dấu hiệu và triệu chứng
1. Kariyawasam D, D'Silva A, Mowat D, et al: Incidence of Duchenne muscular dystrophy in the modern era; an Australian study. Eur J Hum Genet 30(12):1398-1404, 2022. doi: 10.1038/s41431-022-01138-2
2. McDonald DG, Kinali M, Gallagher AC, et al: Fracture prevalence in Duchenne muscular dystrophy. Dev Med Child Neurol 44(10):695-698, 2002. doi: 10.1017/s0012162201002778
3. Duan D, Goemans N, Takeda S, et al: Duchenne muscular dystrophy. Nat Rev Dis Primers 7(1):13, 2021. doi: 10.1038/s41572-021-00248-3
Chẩn đoán
Phân tích đột biến DNA
Sinh thiết cơ với nhuộm miễn dịch của dystrophin
Chẩn đoán được nghi ngờ bởi các biểu hiện lâm sàng đặc trưng, tuổi khi bắt đầu, và tiền sử gia đình gợi ý di truyền lặn liên kết với X. Chẩn đoán được nghi ngờ bởi các biểu hiện lâm sàng đặc trưng, tuổi khi bắt đầu, và tiền sử gia đình gợi ý di truyền lặn liên kết với X. Những thay đổi về bệnh lý cơ được ghi nhận trên điện cơ (điện thế đơn vị vận động xuất hiện nhanh, trong thời gian ngắn, biên độ thấp) và sinh thiết cơ (hoại tử và thể đổi rõ rệt trong kích thước sợi cơ không bị tách rời bởi đơn vị vận động). Nồng độ creatinine kinase tăng lên đến 100 lần bình thường.
Phân tích đột biến DNA từ bạch cầu trong máu ngoại vi bằng cách sử dụng khuếch đại đầu dò phụ thuộc phản ứng nối đa thành phần (MLPA) là xét nghiệm khẳng định chính; nó có thể xác định các bất thường trong gen dystrophin. Nếu khuếch đại đầu dò phụ thuộc phản ứng nối đa thành phần không phát hiện ra bất thường nhưng vẫn nghi ngờ chứng loạn dưỡng Duchenne hoặc Becker, có thể thực hiện giải trình tự đầy đủ của gen dystrophin để phát hiện những thay đổi di truyền nhỏ, chẳng hạn như đột biến điểm.
Nếu xét nghiệm di truyền không xác định chẩn đoán, thì nên phân tích dystrophin với nhuộm miễn dịch các mẫu sinh thiết cơ. Dystrophin không thể phát hiện ở những bệnh nhân bị chứng loạn dưỡng cơ Duchenne. Ở những bệnh nhân bị loạn dưỡng cơ Becker, dystrophin thường là bất thường điển hình (trọng lượng phân tử thấp hơn) hoặc có nồng độ thấp.
Bệnh nhân bị chứng loạn dưỡng cơ Duchenne nên có một đánh giá cơ bản về chức năng tim bằng ECG và siêu âm tim tại thời điểm chẩn đoán hoặc trước 6 tuổi.
Có thể phát hiện người mang gen và chẩn đoán trước khi sinh bằng cách sử dụng các nghiên cứu thông thường (ví dụ, phân tích phả hệ, xác định nồng độ creatinine kinase, xác định giới tính thai nhi) kết hợp với phân tích ADN tái tổ hợp và nhuộm miễn dịch dystrophin của mô cơ.
Điều trị
Các biện pháp hỗ trợ
Có thể phẫu thuật chỉnh hình
Đôi khi, đối với bệnh cơ tim, có thể dùng thuốc ức chế men chuyển angiotensin và/hoặc thuốc chẹn beta
Đối với bệnh loạn dưỡng Duchenne, prednisone hoặc deflazacort và đôi khi là liệu pháp di truyền
Điều trị bao gồm một cách tiếp cận đa ngành bao gồm cả các biện pháp không dùng thuốc và dùng thuốc, bao gồm các liệu pháp di truyền. Tập thể dục tích cực nhẹ nhàng (nghĩa là dưới mức tối đa) được khuyến khích càng lâu càng tốt để tránh tình trạng teo co lại hoặc biến chứng do không hoạt động. Passive exercises may extend the period of ambulation. Các bài tập thụ động có thể kéo dài thời gian vận động Các biện pháp can thiệp chỉnh hình nên nhằm mục đích duy trì chức năng và ngăn ngừa các co rút cơ. Đeo nẹp mắt cá-bàn chân trong giấc ngủ có thể giúp ngăn ngừa sự co rút cơ gấp. Nẹp cẳng chân có thể tạm thời giúp bảo tồn vận động hoặc đứng. Đôi khi cần phải giải phẫu điều trị, đặc biệt đối với chứng vẹo cột sống. Nên tránh bệnh béo phì; các yêu cầu về năng lượng có thể ít hơn bình thường vì hoạt động thể chất giảm.
Suy hô hấp có thể được điều trị bằng hỗ trợ thông khí không xâm lấn (ví dụ: mặt nạ mũi) và đôi khi bằng thở máy. Phẫu thuật mở khí quản tự chọn đang được chấp nhận, cho phép trẻ mắc chứng loạn dưỡng Duchenne sống đến độ tuổi 30 trở lên.
Đối với trẻ mắc bệnh cơ tim giãn, thuốc ức chế men chuyển angiotensin và/hoặc thuốc chẹn beta có thể giúp ngăn ngừa hoặc làm chậm sự tiến triển (1).
Các liệu pháp nghiên cứu chứng loạn dưỡng Duchenne và chứng loạn dưỡng Becker bao gồm liệu pháp gen, creatine, bất hoạt myostatin, chất tạo ra cơ xương và chất chống oxy hóa idebenone (2).
Tư vấn di truyền được chỉ định.
Corticosteroid điều trị chứng loạn dưỡng Duchenne
Trong bệnh loạn dưỡng Duchenne, corticosteroid hàng ngày (prednisone hoặc deflazacort) là phương pháp điều trị chính cho bệnh nhân > 4 tuổi không còn phát triển hoặc suy giảm các kỹ năng vận động (3). Corticosteroid bắt đầu có tác dụng sớm nhất là 10 ngày sau khi bắt đầu điều trị; hiệu quả đạt đỉnh điểm ở thời điểm 3 tháng và kéo dài trong 6 tháng. Sử dụng lâu dài giúp cải thiện sức mạnh, trì hoãn độ tuổi mất khả năng đi lại từ 1,4 tuổi đến 2,5 tuổi, cải thiện việc kiểm tra chức năng theo thời gian (đo lường tốc độ trẻ hoàn thành một nhiệm vụ chức năng, chẳng hạn như đi bộ hoặc đứng dậy từ sàn), cải thiện chức năng phổi, làm giảm các biến chứng do chỉnh hình (ví dụ: việc cần phải phẫu thuật điều trị vẹo cột sống), ổn định chức năng tim (ví dụ: trì hoãn khởi phát bệnh cơ tim cho đến 18 tuổi) và tăng thời gian sống thêm từ 5 năm đến 15 năm (3). Sử dụng prednisone cách nhật không hiệu quả. Tăng cân và bộ mặt Cushing là những tác dụng phụ thường gặp sau 6 đến 18 tháng. Nguy cơ xẹp cột sống cũng như xương gãy xương cũng tăng lên.
Deflazacort có thể liên quan đến nguy cơ đục thủy tinh thể cao hơn prednisone.
Việc sử dụng prednisone hoặc deflazacort trong bệnh loạn dưỡng Becker chưa được nghiên cứu đầy đủ.
Các lựa chọn liệu pháp di truyền cho bệnh loạn dưỡng Duchenne
Các liệu pháp di truyền làm tăng nồng độ dystrophin đã có sẵn ở một số quốc gia, nhưng các liệu pháp này rất tốn kém và lợi ích không chắc chắn (4). Việc sử dụng các liệu pháp này đòi hỏi phải cân nhắc cẩn thận và đưa ra quyết định chung.
Các thuốc loại bỏ exon (eteplirsen, golodirsen, viltolarsen và casimersen đường tĩnh mạch) sử dụng các oligonucleotide antisense hoạt động giống như các miếng vá phân tử đối với gen dystrophin bất thường trong đó thiếu 1 hoặc nhiều exon (các exon bị thiếu sẽ ngăn cản quá trình lắp ráp protein hoàn chỉnh, do đó gây ra các triệu chứng nặng). Những loại thuốc này che giấu một exon để nó bị bỏ qua và bị bỏ qua trong quá trình sản sinh protein, cho phép sản sing protein dystrophin, mặc dù không bình thường nhưng có chức năng và về mặt lý thuyết là làm giảm các triệu chứng để bệnh nhân hoạt động giống như những cậu bé mắc chứng loạn dưỡng cơ Becker nhẹ hơn.
Eteplirsen bỏ qua exon 51. Dữ liệu hạn chế cho thấy eteplirsen dẫn đến tăng dystrophin trong cơ và tăng hiệu suất đi bộ trong các bài kiểm tra tính thời gian ở 13% số bệnh nhân mắc chứng loạn dưỡng Duchenne có đột biến gen dystrophin, đột biến này có thể bỏ qua exon 51. Việc phê duyệt thuốc đã bị chỉ trích vì nó dựa trên một thử nghiệm nhỏ dựa vào kết quả thay thế (dystrophin trong sinh thiết cơ) (5), và lợi ích lâm sàng vẫn chưa được chứng minh.
Golodirsen và viltolarsen bỏ qua exon 53. Các thuốc có thể được sử dụng cho 8% số bệnh nhân mắc chứng loạn dưỡng Duchenne có đột biến gen dystrophin có thể bỏ qua exon 53. Lợi ích lâm sàng vẫn chưa được chứng minh.
Casimersen bỏ qua exon 45. Thuốc này có thể được sử dụng ở 8% số bệnh nhân mắc chứng loạn dưỡng Duchenne có đột biến được xác nhận có thể bỏ qua exon 45. Thuốc làm tăng sản sinh dystrophin, nhưng lợi ích lâm sàng chưa được chứng minh.
Thuốc đọc qua codon dừng (ví dụ: ataluren đường uống [PTC124]) bỏ qua các codon dừng sớm, cho phép sản sinh protein chức năng. Codon dừng là những đột biến vô nghĩa làm ngừng quá trình sản sinh protein hoạt động quá sớm, dẫn đến protein bị cắt bớt, không có chức năng.
Ataluren thúc đẩy quá trình đọc qua ribosome của các codon kết thúc sớm nhưng không bình thường và nhằm mục đích tạo ra protein dystrophin chức năng. Đây là một phương án dành cho những bệnh nhân mắc chứng loạn dưỡng Duchenne từ 2 tuổi trở lên, có khả năng đi lại được và bệnh do đột biến vô nghĩa gây ra. Ataluren có sẵn ở Liên minh Châu Âu và Vương quốc Anh. Lợi ích lâm sàng chưa được chứng minh (6).
Chuyển gen thông qua các vec tơ vi rút (ví dụ: delandistrogene moxeparvovec) sử dụng các vec tơ vi rút để đưa vật liệu di truyền điều chỉnh đến các cơ bị ảnh hưởng.
Mặc dù chưa chứng minh được lợi ích lâm sàng, nhưng delandistrogene moxeparvovec là thuốc duy nhất được phê duyệt cho liệu pháp chuyển gen thông qua vec tơ vi rút, nhờ đó các gen chuyển microdystrophin có thể được chuyển đến cơ xương và cơ tim bằng cách sử dụng capsid vi rút liên quan đến adeno (AAVrh74). Không thấy sự cải thiện chức năng nào trong thử nghiệm (7), nhưng trong phân tích phân nhóm, một số cải thiện về tình trạng chức năng đã được quan sát thấy ở nhóm tuổi từ 4 tuổi đến 5 tuổi (8). Các tác dụng bất lợi bao gồm buồn nôn, nôn, sốt, rối loạn chức năng gan và giảm tiểu cầu cũng như các phản ứng viêm miễn dịch đe dọa tính mạng. Cần nghiên cứu sâu hơn để xác định vị trí của thuốc này trong vũ khí trị liệu cho chứng loạn dưỡng Duchenne.
Tài liệu tham khảo về điều trị
1. Birnkrant DJ, Bushby K, Bann CM, et al: Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 17(4):347-361, 2018. doi: 10.1016/S1474-4422(18)30025-5
2. Ren S, Yao C, Liu Y, et al: Antioxidants for Treatment of Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis. Eur Neurol 85(5):377-388, 2022. doi: 10.1159/000525045
3. Gloss D, Moxley RT 3rd, Ashwal S, Oskoui M: Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 86:465–472, 2016. doi: 10.1212/WNL.0000000000002337
4. Bendicksen L, Zuckerman DM, Avorn J, et al: The Regulatory Repercussions of Approving Muscular Dystrophy Medications on the Basis of Limited Evidence. Ann Intern Med 176(9):1251-1256, 2023. doi: 10.7326/M23-1073
5. Mendell JR, Goemans N, Lowes LP, et al: Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79(2):257-271, 2016. doi: 10.1002/ana.24555
6. McDonald CM, Campbell C, Torricelli RE, et al: Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390:(10101):1489–1498, 2017. doi: 10.1016/S0140-6736(17)31611-2
7. Mendell JR, Shieh PB, McDonald CM, et al: Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy. Front Cell Dev Biol 11:1167762, 2023. doi: 10.3389/fcell.2023.1167762
8. Elevidys. Prescribing information. Sarepta Therapeutics, Inc; 2023. Truy cập ngày 9 tháng 1 năm 2024.
Những điểm chính
Chứng loạn dưỡng Duchenne và chứng Becker là các bệnh di truyền lặn liên kết X gây giảm dystrophin, một protein trong màng tế bào cơ.
Bệnh nhân bị yếu cơ đáng kể, tiến triển gây ra tàn tật trầm trọng, bao gồm khó vận động, ngã thường xuyên, bệnh cơ tim giãn, và tử vong sớm do suy hô hấp.
Tập thể dục chủ động và thụ động thì hữu ích, cùng với nẹp chân và dụng cụ chỉnh hình mắt cá - bàn chân.
Trong chứng loạn dưỡng Duchenne, prednisone hoặc deflazacort hàng ngày giúp cải thiện cơ lực và khối lượng cơ, cải thiện chức năng phổi và giúp trì hoãn sự khởi phát của bệnh cơ tim, mặc dù các tác dụng bất lợi thường gặp.
Đối với những bệnh nhân mắc chứng loạn dưỡng Duchenne có đột biến nhất định, eteplirsen, golodirsen, viltolarsen, casimersen và ataluren, mặc dù có bằng chứng hạn chế về lợi ích lâm sàng, cũng có thể được sử dụng.
Thuốc ức chế men chuyển angiotensin và/hoặc thuốc chẹn beta có thể giúp ngăn ngừa hoặc làm chậm sự tiến triển của bệnh cơ tim.
Hỗ trợ thở máy (không xâm lấn và, sau này, xâm lấn) có thể giúp kéo dài cuộc sống.
Thông tin thêm
Sau đây là các tài nguyên tiếng Anh có thể hữu ích. Vui lòng lưu ý rằng CẨM NANG không chịu trách nhiệm về nội dung của các tài nguyên này.
Muscular Dystrophy Association: Information on research, treatment, technology, and support for patients living with Duchenne muscular dystrophy and Becker muscular dystrophy
National Organization for Rare Disorders: Comprehensive information regarding Duchenne muscular dystrophy and Becker muscular dystrophy, including standard and investigational therapies and links to related topics
Muscular Dystrophy News Today: A news and information web site about muscular dystrophy


