


- Overview of Interstitial Lung Disease
- Drug-Induced Pulmonary Disease
- Overview of Eosinophilic Pulmonary Diseases
- Acute Eosinophilic Pneumonia
- Chronic Eosinophilic Pneumonia
- Löffler Syndrome
- Hypersensitivity Pneumonitis
- Overview of Idiopathic Interstitial Pneumonias
- Idiopathic Pulmonary Fibrosis (IPF)
- Desquamative Interstitial Pneumonia (DIP)
- Nonspecific Interstitial Pneumonia (NSIP)
- Cryptogenic Organizing Pneumonia (COP)
- Respiratory Bronchiolitis–Associated Interstitial Lung Disease (RBILD)
- Acute Interstitial Pneumonia (AIP)
- Lymphoid Interstitial Pneumonia (LIP)
- Idiopathic Pleuroparenchymal Fibroelastosis
- Lymphangioleiomyomatosis
- Pulmonary Alveolar Proteinosis
- Pulmonary Langerhans Cell Histiocytosis
Pulmonary alveolar proteinosis is accumulation of surfactant in alveoli. Etiology is almost always unknown. Symptoms are dyspnea, fatigue, and malaise. Diagnosis is based on bronchoalveolar lavage, although characteristic radiograph and laboratory test abnormalities occur. Treatment is with whole lung lavage or, in some cases, recombinant granulocyte-macrophage colony stimulating factor. Five-year survival is approximately 80% with treatment.
")
Etiology of Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis is most often idiopathic and occurs in otherwise healthy men and women between 30 and 50 years. There is thought to be a male preponderance and an association with cigarette smoking (1). The population prevalence is estimated to be around 7 to 10 per million (2). Rare secondary forms occur in patients with:
Hematologic cancers
Immunosuppression by medications
Significant inhalation exposures to aluminum, titanium, cement, and cellulose dusts
Rare congenital forms that cause neonatal respiratory failure also exist.
It is unclear whether idiopathic and secondary cases share a common pathophysiology.
Etiology references
1. Trapnell BC, Nakata K, Bonella F, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers 2019;5(1):16. doi:10.1038/s41572-019-0066-3
2. McCarthy C, Carey BC, Trapnell BC. Autoimmune Pulmonary Alveolar Proteinosis. Am J Respir Crit Care Med 2022;205(9):1016-1035. doi:10.1164/rccm.202112-2742SO
Pathophysiology of Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis is thought to be due to disorders of surfactant clearance or production. Impaired alveolar macrophage processing of surfactant due to abnormal granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling is thought to contribute to the disorder, perhaps due to reduced or absent function of the common beta chain of the GM-CSF/interleukin (IL)-13/IL-5 receptor on mononuclear cells (present in some children but not in adults with the disorder). Anti–GM-CSF antibodies have also been found in most patients. Toxic lung injury is suspected but not proved in secondary inhalation causes, such as silicoproteinosis.
Alveoli are filled with acellular lipoprotein surfactant that stains periodic acid–Schiff (PAS) positive. Alveolar and interstitial cells remain normal. Posterobasal lung segments are mostly affected. The pleura and mediastinum are unaffected.
Symptoms and Signs of Pulmonary Alveolar Proteinosis
Most patients with pulmonary alveolar proteinosis present with progressive exertional dyspnea and weight loss, fatigue, malaise, or low-grade fever. Cough, occasionally producing chunky or gummy sputum, occurs but is less common. Clubbing and cyanosis are uncommon. Inspiratory crackles are rare because alveoli are fluid-filled; when crackles are present, they suggest infection.
Diagnosis of Pulmonary Alveolar Proteinosis
Chest radiograph and high-resolution CT (HRCT)
Bronchoalveolar lavage
Sometimes biopsy
Pulmonary alveolar proteinosis is usually first suspected when a chest radiograph is taken for nonspecific respiratory symptoms. The radiograph shows bilateral mid- and lower-lung field opacities in a butterfly distribution with normal hila.

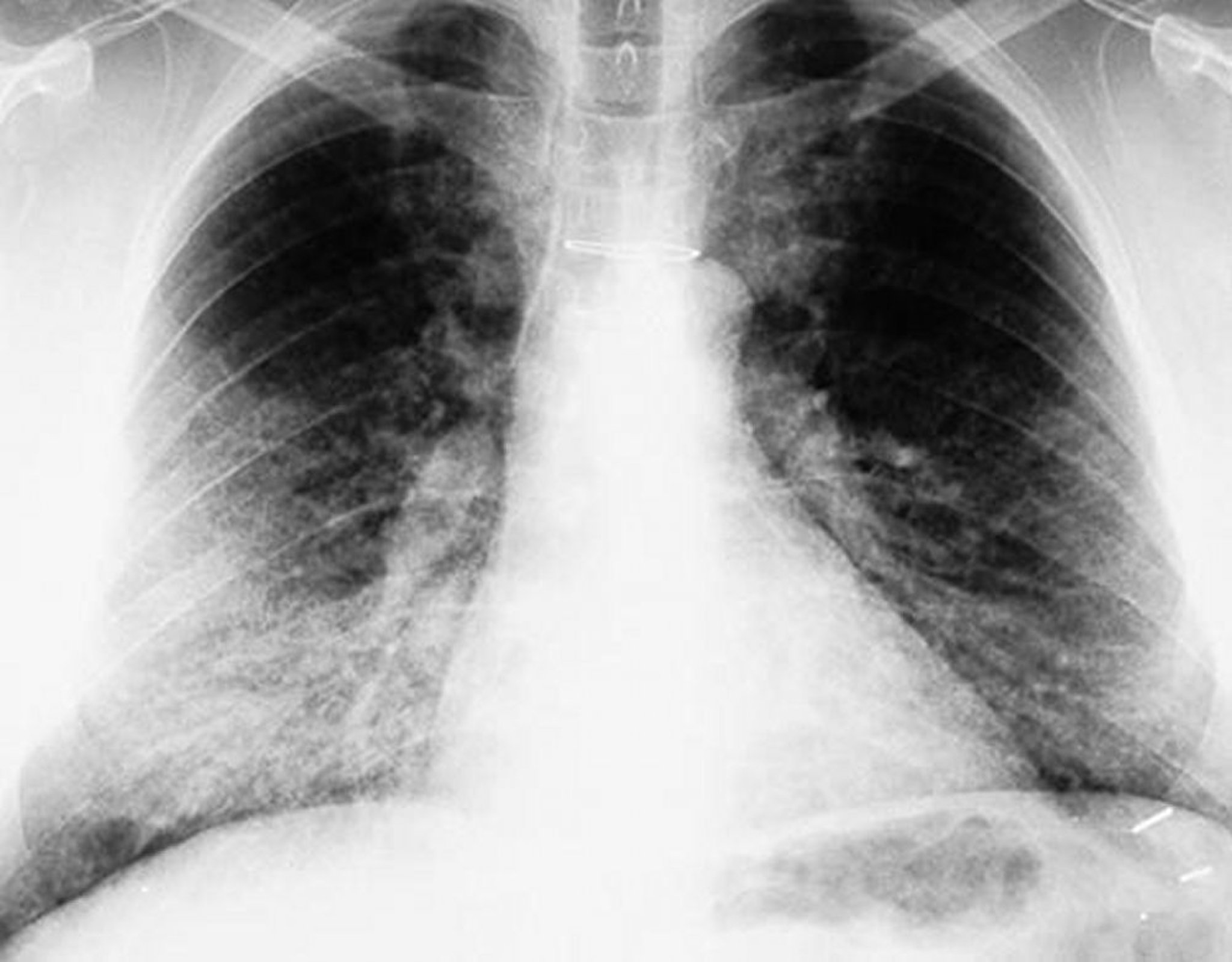
By permission of the publisher. From Lynch III J, Myers J. In Bone's Atlas of Pulmonary and Critical Care Medicine. Edited by J Crapo. Philadelphia, Current Medicine, 2005.
Bronchoalveolar lavage is done. Lavage fluid is milky or opaque and stains positive with periodic acid–Schiff (PAS) staining. Lavage fluid is characterized by scattered surfactant-engorged macrophages, an increase in T cells, and high levels of surfactant apoprotein-A.
Thoracoscopic or open lung biopsy is done when bronchoscopy is contraindicated or when specimens from lavage fluid are nondiagnostic. Tests typically done before treatment begins include
Arterial blood gas measurements (ABGs)
Laboratory tests
Pulmonary function tests
ABGs may show hypoxemia with mild to moderate exercise or at rest if disease is more severe.
HRCT shows ground-glass opacification, thickened intralobular structures, and interlobular septa in typical polygonal shapes (crazy-paving). This finding is not specific, however, as it may also occur in patients with acute respiratory distress syndrome (ARDS), viral pneumonia, lipoid pneumonia, bronchoalveolar cell carcinoma, and Pneumocystis jirovecii pneumonia.

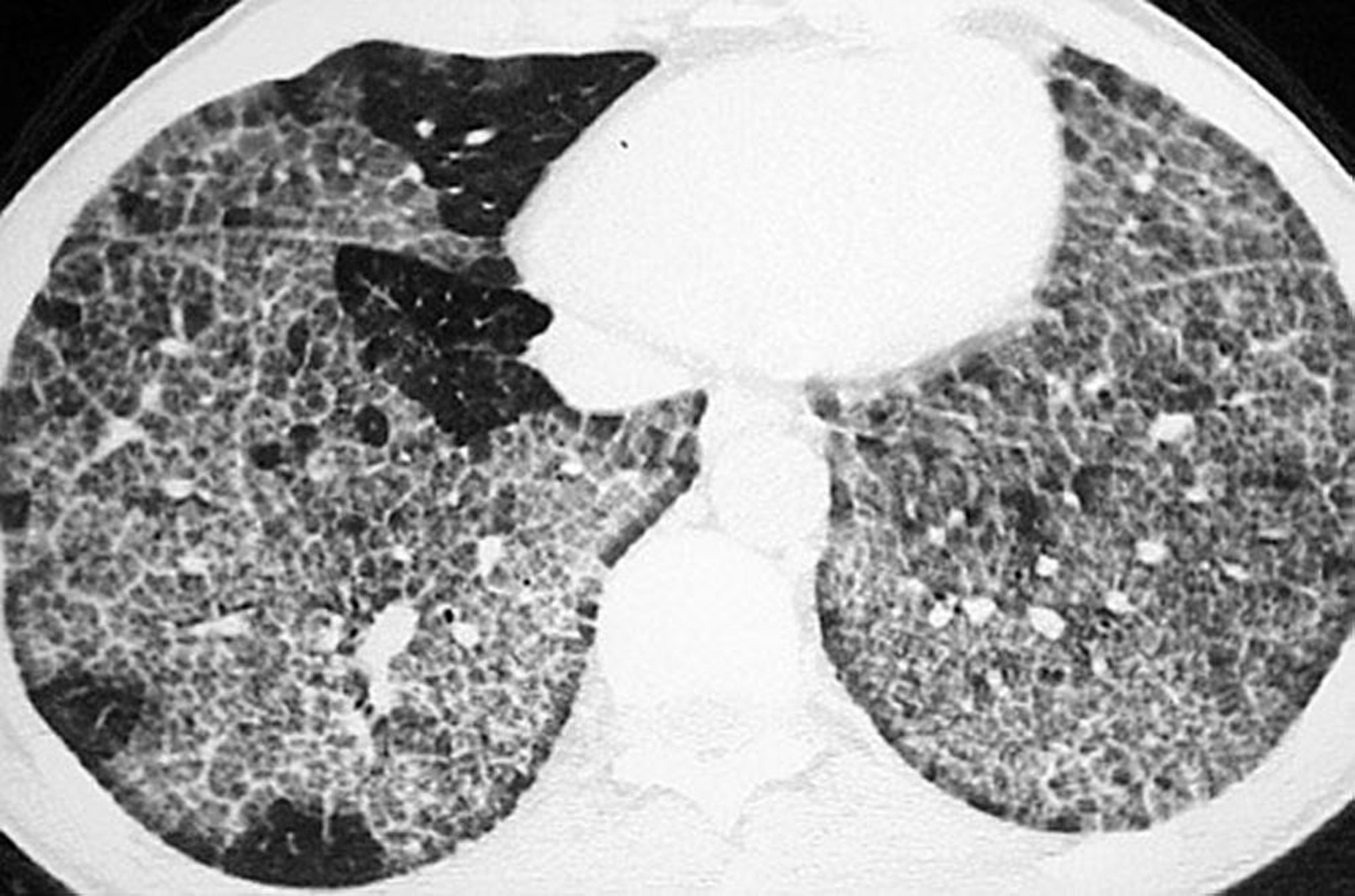
Image courtesy of Talmadge E. King, MD.
Pulmonary function tests show reduction in diffusing capacity of the lung for carbon monoxide (DLCO) that is disproportionate to the decreases in vital capacity, residual volume, functional residual capacity, and total lung capacity.
Laboratory test abnormalities include polycythemia, hypergammaglobulinemia, increased serum lactate dehydrogenase levels, and increased serum surfactant proteins A and D. Abnormalities are suggestive but nondiagnostic. Laboratory tests for autoantibodies to granulocyte-macrophage colony-stimulating factor (GM-CSF) can help support an autoimmune etiology to pulmonary alveolar proteinosis.
Treatment of Pulmonary Alveolar Proteinosis
Whole lung lavage
Treatment of pulmonary alveolar proteinosis is unnecessary for patients without symptoms or for those with only mild symptoms.
In patients with troubling dyspnea, whole lung lavage is done by using general anesthesia and a double-lumen endotracheal tube. Lavage of one lung is done up to 15 times with 1 to 2 L saline while the other lung is ventilated. The process is then reversed.
Lung transplantation is not often done because the disorder recurs in the transplanted lung.
Systemic glucocorticoids play no role in management and may increase the risk of secondary infection. The role of GM-CSF (inhaled or subcutaneous) in routine management has not been determined, but it may be of benefit in selected cases. A multicenter, self-controlled, phase 2 trial at nine pulmonary centers in Japan showed an overall response rate of 62% (24/39) (1).
Treatment reference
1. Tazawa R, Trapnell BC, Inoue Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med 2010;181(12):1345-1354. doi:10.1164/rccm.200906-0978OC
Prognosis for Pulmonary Alveolar Proteinosis
Without treatment, pulmonary alveolar proteinosis remits spontaneously in up to 10% of patients (1). When patients are treated with whole lung lavage, the 5-year survival rate is 95% (1).
Secondary pulmonary infections with bacterial (eg, Mycobacteria, Nocardia) and other organisms (eg, Aspergillus, Cryptococcus, and other opportunistic fungi) occasionally develop because of impaired macrophage function; these infections require treatment.
Prognosis reference
1. Campo I, Mariani F, Rodi G, et al. Assessment and management of pulmonary alveolar proteinosis in a reference center. Orphanet J Rare Dis 2013;8:40. doi:10.1186/1750-1172-8-40
Key Points
Consider pulmonary alveolar proteinosis in otherwise healthy patients between ages 30 and 50 years if the chest radiograph shows bilateral mid- and lower-lung field opacities in a butterfly distribution with normal hila.
Perform bronchoalveolar lavage and laboratory tests; if contraindicated or when results are not diagnostic, perform a lung biopsy.
If dyspnea is moderate or severe, treat with whole lung lavage.







