


- Overview of Interstitial Lung Disease
- Drug-Induced Pulmonary Disease
- Overview of Eosinophilic Pulmonary Diseases
- Acute Eosinophilic Pneumonia
- Chronic Eosinophilic Pneumonia
- Löffler Syndrome
- Hypersensitivity Pneumonitis
- Overview of Idiopathic Interstitial Pneumonias
- Idiopathic Pulmonary Fibrosis (IPF)
- Desquamative Interstitial Pneumonia (DIP)
- Nonspecific Interstitial Pneumonia (NSIP)
- Cryptogenic Organizing Pneumonia (COP)
- Respiratory Bronchiolitis–Associated Interstitial Lung Disease (RBILD)
- Acute Interstitial Pneumonia (AIP)
- Lymphoid Interstitial Pneumonia (LIP)
- Idiopathic Pleuroparenchymal Fibroelastosis
- Lymphangioleiomyomatosis
- Pulmonary Alveolar Proteinosis
- Pulmonary Langerhans Cell Histiocytosis
Lymphangioleiomyomatosis (LAM) is an indolent, progressive growth of smooth muscle cells throughout the lungs, pulmonary blood vessels, lymphatics, and pleurae. It is rare and occurs mostly in young women. Symptoms are dyspnea, cough, chest pain, and hemoptysis; chylous effusions and spontaneous pneumothorax are common. Diagnosis is suspected on the basis of symptoms and chest radiograph findings and is confirmed by high-resolution CT. Treatment is with sirolimus or lung transplantation. Prognosis is uncertain, but the disorder is slowly progressive and over years often leads to respiratory failure and death.
Topic Resources

Lymphangioleiomyomatosis (LAM) is a rare disease typically affecting women between 20 and 40 years. White women are at greatest risk. LAM affects < 1 in 1 million people per year (1); however, with advances in screening, the prevalence is increasing (2).
Lymphangioleiomyomatosis is characterized by proliferation of atypical smooth muscle cells throughout the chest, including lung parenchyma, vasculature, lymphatics, and pleurae, leading to distortion of lung architecture, cystic emphysema, and progressive deterioration of lung function.
General references
1. Harknett EC, Chang WY, Byrnes S, et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM 2011;104(11):971-979. doi:10.1093/qjmed/hcr116
2. Lynn E, Forde SH, Franciosi AN, et al. Updated Prevalence of Lymphangioleiomyomatosis in Europe. Am J Respir Crit Care Med 2024;209(4):456-459. doi:10.1164/rccm.202310-1736LE
Etiology of Lymphangioleiomyomatosis
The cause of lymphangioleiomyomatosis is thought to be due to loss-of-function genetic mutations of the tuberous sclerosis proteins, tuberin and hamartin. Unregulated expression of growth factors have been implicated in the pathogenesis. The hypothesis that female sex hormones play a role in pathogenesis remains unproven.
The disease usually arises spontaneously, but LAM bears many similarities to the pulmonary findings of tuberous sclerosis; LAM occurs in some patients with tuberous sclerosis and is thought by some to be a forme fruste of tuberous sclerosis. Mutations in the tuberous sclerosis complex-2 gene (TSC-2) have been described in LAM cells and angiomyolipomas (benign renal hamartomas made of smooth muscle, blood vessels, and adipose). Angiomyolipomas occur in up to 50% of patients with LAM. These observations suggest 1 of 2 possibilities:
Somatic mosaicism for TSC-2 mutations within the lungs and kidneys results in foci of disease superimposed against a background of normal cells within these tissues (although multiple discrete sites of disease might be expected). These mutations lead to an unregulated overexpression of growth factors (eg, vascular endothelial growth factor [VEGF], insulin-like growth factor [IGF], platelet-derived growth factor [PDGF], fibroblast growth factor [FGF])which are also thought to contribute to disease progression. Matrix metalloproteinases and cathepsins may be responsible for cell migrations and differentiation, contributing to tissue remodeling.
Lymphangioleiomyomatosis represents a low-grade, destructive, metastasizing neoplasm, perhaps of uterine origin, that spreads through the lymphatic system.
Symptoms and Signs of Lymphangioleiomyomatosis
Initial symptoms of lymphangioleiomyomatosis are fatigue and dyspnea (especially during exertion). These symptoms can be misconstrued as presenting symptoms of asthma or chronic obstructive pulmonary disease (COPD). Less commonly, cough, chest pain, and hemoptysis may occur. There are few signs of disease, but some women have crackles and rhonchi. Rarely, pulmonary hypertension occurs.
Many patients present with spontaneous pneumothorax as well as pleural effusions. They may also present with manifestations of axial lymphadenopathy, often causing lymphatic obstruction, including chylothorax, chylous ascites, and chyluria. Symptoms are thought to worsen during pregnancy.
Renal angiomyolipomas, although usually asymptomatic, can cause bleeding if they grow large (eg, > 4 cm), usually manifesting as hematuria or flank pain.
Diagnosis of Lymphangioleiomyomatosis
Chest radiograph and high-resolution CT (HRCT)
Pulmonary function testing
VEGF-D testing
Lung biopsy if HRCT and VEGF-D testing are nondiagnostic
Diagnosis of lymphangioleiomyomatosis is suspected in young women with dyspnea plus interstitial changes with normal or increased lung volumes on chest radiograph, spontaneous pneumothorax, or chylous effusion (1). Lymphangioleiomyomatosis is often misdiagnosed as another form of interstitial lung disease.
HRCT is recommended in all patients suspected of having the disorder; findings of multiple, small, diffusely distributed, thin-walled cysts are generally pathognomonic for LAM. Pneumothorax,pleural effusions, and chylous effusions (in the thorax or abdomen) may be seen on imaging.

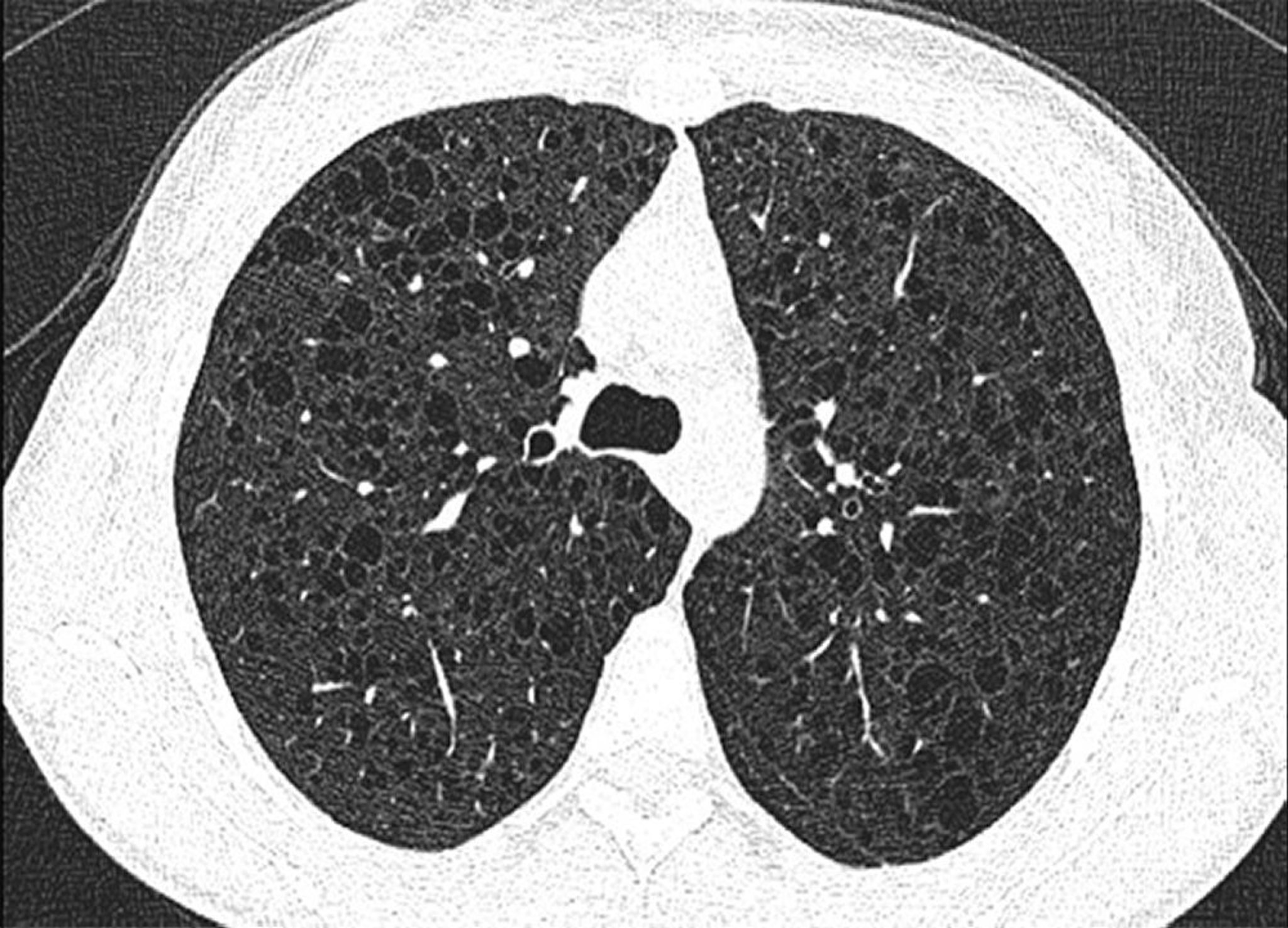
Image courtesy of Joyce Lee, MD, MAS.
Serum VEGF-D (vascular endothelial growth factor D) testing is recommended. Serum VEGF-D levels are elevated in the majority of women with LAM and are normal in women with other forms of cystic lung disease. An elevated VEGF-D level can confirm LAM, but a normal level does not exclude the diagnosis (2).
Biopsy (surgical) is indicated only when HRCT findings and VEGF-D testing are nondiagnostic. Findings of an abnormal proliferation of smooth muscle cells (LAM cells) associated with cystic changes on histologic examination confirm disease.
Pulmonary function tests support the diagnosis and are especially useful for monitoring. Typical findings are of an obstructive or mixed obstructive and restrictive pattern. The lungs are usually hyperinflated with an increase in the total lung capacity (TLC) and thoracic gas volume. Gas trapping (an increase in residual volume [RV] and RV/TLC ratio) is commonly present. The partial pressure of arterial oxygen (PaO2) and diffusing capacity of the lung for carbon monoxide (DLCO) are commonly reduced. Exercise performance is decreased in most patients.
Diagnosis references
1. McCarthy C, Gupta N, Johnson SR, Yu JJ, McCormack FX. Lymphangioleiomyomatosis: pathogenesis, clinical features, diagnosis, and management. Lancet Respir Med 2021;9(11):1313-1327. doi:10.1016/S2213-2600(21)00228-9
2. McCormack FX, Gupta N, Finlay GR, et al: Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med 194 (6):748–761, 2016.
Treatment of Lymphangioleiomyomatosis
Sirolimus Sirolimus
Lung transplantation
Sirolimus (an mTOR inhibitor) may help stabilize or slow the decline in pulmonary function among patients with moderate lung impairment (forced expiratory volume in 1 second [FEV1] < 70% predicted). Treatment withSirolimus (an mTOR inhibitor) may help stabilize or slow the decline in pulmonary function among patients with moderate lung impairment (forced expiratory volume in 1 second [FEV1] < 70% predicted). Treatment withsirolimus is recommended for patients with abnormal or declining lung function (1). Alternative treatments, such as hormonal manipulation with progestins, tamoxifen, oophorectomy, or anti-inflammatory tetracycline therapy are largely ineffective and not recommended. ). Alternative treatments, such as hormonal manipulation with progestins, tamoxifen, oophorectomy, or anti-inflammatory tetracycline therapy are largely ineffective and not recommended.
Pneumothorax may be difficult to manage because it often recurs, is bilateral, and is less responsive to standard measures. Recurrent pneumothorax requires pleural abrasion, talc or chemical pleurodesis, or pleurectomy. Embolization to prevent bleeding should be considered for angiomyolipomas Pneumothorax may be difficult to manage because it often recurs, is bilateral, and is less responsive to standard measures. Recurrent pneumothorax requires pleural abrasion, talc or chemical pleurodesis, or pleurectomy. Embolization to prevent bleeding should be considered for angiomyolipomas> 4 cm.
Standard treatment of lymphangioleiomyomatosis is lung transplantation, but the disorder can recur in transplanted lungs.
Air travel is well-tolerated by most patients (2).
Treatment references
1. Feemster LC, Lyons PG, Chatterjee RS, et al. Summary for Clinicians: Lymphangioleiomyomatosis Diagnosis and Management Clinical Practice Guideline. Ann Am Thorac Soc 2017;14(7):1073-1075. doi:10.1513/AnnalsATS.201609-685CME
2. Taveira-DaSilva AM, Burstein D, Hathaway OM, et al. Pneumothorax after air travel in lymphangioleiomyomatosis, idiopathic pulmonary fibrosis, and sarcoidosis. Chest 2009;136(3):665-670. doi:10.1378/chest.08-3034
Prognosis for Lymphangioleiomyomatosis
The prognosis for patients with lymphangioleiomyomatosis is unclear because the disorder is so rare and because the clinical course of patients with LAM is variable. In general, the disease is slowly progressive, leading eventually to respiratory failure and death. The estimated 10-year survival rates are variable, but exceed 90% and are more favorable than previously believed (1). Women should be advised that progression may accelerate during pregnancy.
Prognosis reference
1. Gupta N, Lee HS, Ryu JH, et al. The NHLBI LAM Registry: Prognostic Physiologic and Radiologic Biomarkers Emerge From a 15-Year Prospective Longitudinal Analysis. Chest 2019;155(2):288-296. doi:10.1016/j.chest.2018.06.016
Key Points
Lymphangioleiomyomatosis (LAM) can mimic interstitial lung disease but is actually a rare, slowly progressive growth of smooth muscle cells in various organs, including the lungs.
Consider the diagnosis in young women with unexplained dyspnea plus interstitial changes with normal or increased lung volumes on chest radiograph, spontaneous pneumothorax, or chylous pleural effusion.
Perform high-resolution CT and VEGF-D (vascular endothelial growth factor D) testing; if results are inconclusive, consider a surgical lung biopsy.
Consider sirolimus treatment for LAM with abnormal or progressively decreasing pulmonary function.Consider sirolimus treatment for LAM with abnormal or progressively decreasing pulmonary function.
More Information
The following English-language resource may be useful. Please note that The Manual is not responsible for the content of this resource.







