



- Overview of Interstitial Lung Disease
- Drug-Induced Pulmonary Disease
- Overview of Eosinophilic Pulmonary Diseases
- Acute Eosinophilic Pneumonia
- Chronic Eosinophilic Pneumonia
- Löffler Syndrome
- Hypersensitivity Pneumonitis
- Overview of Idiopathic Interstitial Pneumonias
- Idiopathic Pulmonary Fibrosis (IPF)
- Desquamative Interstitial Pneumonia (DIP)
- Nonspecific Interstitial Pneumonia (NSIP)
- Cryptogenic Organizing Pneumonia (COP)
- Respiratory Bronchiolitis–Associated Interstitial Lung Disease (RBILD)
- Acute Interstitial Pneumonia (AIP)
- Lymphoid Interstitial Pneumonia (LIP)
- Idiopathic Pleuroparenchymal Fibroelastosis
- Lymphangioleiomyomatosis
- Pulmonary Alveolar Proteinosis
- Pulmonary Langerhans Cell Histiocytosis
Lymphoid interstitial pneumonia (LIP) is lymphocytic infiltration of the alveolar interstitium and air spaces. The cause is unknown. Symptoms and signs are cough, progressive dyspnea, and crackles. Diagnosis is based on history, physical examination, imaging tests, and lung biopsy. Treatment is with glucocorticoids and/or cytotoxic drugs. Prognosis is largely unknown and limited to case-series.
Topic Resources

Lymphoid interstitial pneumonia is a rare idiopathic interstitial pneumonia characterized by infiltration of alveoli and alveolar septa with small lymphocytes and varying numbers of plasma cells. Non-necrotizing, poorly formed granulomas may be present but are usually rare and inconspicuous.
Lymphoid interstitial pneumonia is the most common cause of pulmonary disease after Pneumocystis infection in HIV-positive children and is the late stage HIV infection-defining illness in up to one-half of HIV-positive children. Lymphoid interstitial pneumonia affects < 1% of adults with or without HIV infection. Women and girls are affected more commonly.
The cause is postulated to be an autoimmune disease or a nonspecific response to infection with Epstein-Barr virus, HIV, or other viruses. Evidence of an autoimmune etiology includes its frequent association with Sjögren syndrome and other disorders (eg, systemic lupus erythematosus, rheumatoid arthritis, Hashimoto thyroiditis).
Evidence of an indirect viral etiology includes frequent association with immunodeficient states (HIV, combined variable immunodeficiency, agammaglobulinemia) and findings of Epstein-Barr virus DNA and HIV RNA in lung tissue of patients with lymphoid interstitial pneumonia. According to this theory, lymphoid interstitial pneumonia is an extreme manifestation of the normal ability of lymphoid tissue in the lung to respond to inhaled and circulating antigens.
Most forms of lymphoid interstitial pneumonia are classified as secondary when they occur in association with other conditions (immune, infectious, or drug-induced). When no cause can be identified, it is classified as primary lymphoid interstitial pneumonia.
Symptoms and Signs of Lymphoid Interstitial Pneumonia
In adults, lymphoid interstitial pneumonia typically causes progressive dyspnea and cough. These manifestations progress over months or, in some cases, years and appear at a mean age of 47 years (1). Weight loss, fever, arthralgias, pleuritic chest pain, and night sweats occur but are less common.
Examination may reveal crackles. Findings such as hepatosplenomegaly, arthritis, and lymphadenopathy are uncommon and suggest an accompanying or alternative diagnosis (eg, lymphoma).
Symptoms and signs reference
1. Cha SI, Fessler MB, Cool CD, Schwarz MI, Brown KK. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J 2006;28(2):364-369. doi:10.1183/09031936.06.00076705
Diagnosis of Lymphoid Interstitial Pneumonia
High-resolution CT (HRCT)
Laboratory testing for identification of underlying disorder
Pulmonary function tests
For confirmation, biopsy
Diagnosis of lymphoid interstitial pneumonia is usually suspected in at-risk patients with compatible symptoms. Imaging tests and sometimes lung biopsy are performed.
Chest radiograph shows bibasilar linear reticular or nodular opacities, a nonspecific finding that is present in a number of pulmonary infections. Alveolar opacities, cysts, or both may be present in more advanced disease.
HRCT of the chest should be performed to establish the extent of disease, define the hilar anatomy, and identify pleural involvement. HRCT findings are highly variable. Characteristic findings are centrilobular and subpleural nodules, thickened bronchovascular bundles, nodular ground-glass opacities, and cystic structures.
Pulmonary function testing reveals a restrictive lung disease pattern with evidence of reduced lung volumes (total lung capacity and forced vital capacity) and reduced diffusing capacity of the lung for carbon dioxide (DLCO).

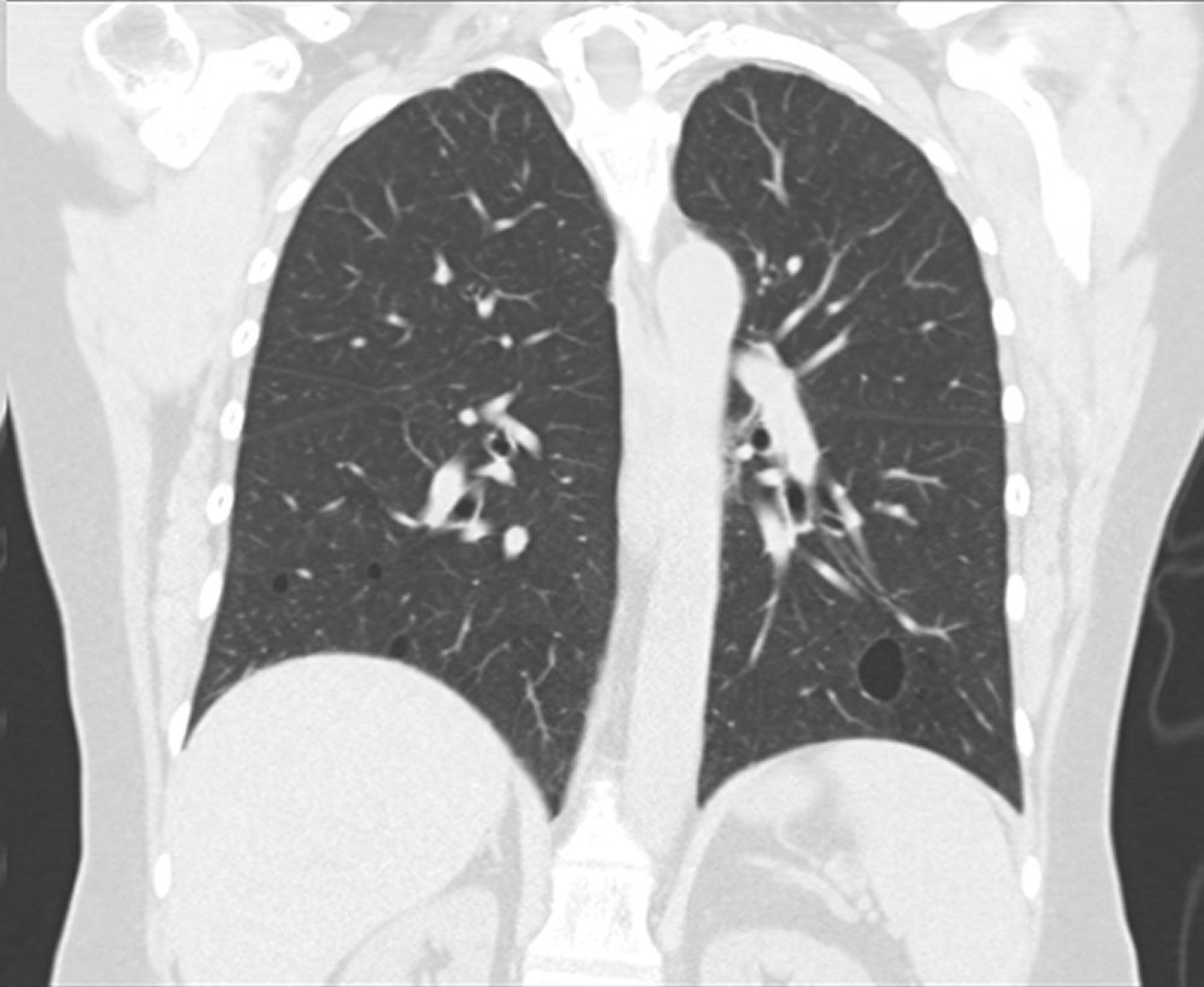
This image courtesy of Tami Bang, MD.
Marked hypoxemia may occur.
Bronchoalveolar lavage should be done to exclude infection and may reveal an increased number of lymphocytes.
Routine laboratory testing and serum protein electrophoresis (SPEP) are done because approximately 80% of patients can have a dysproteinemia (serum protein abnormality), most commonly a polyclonal gammopathy or hypogammaglobulinemia, the significance of which is unknown (1). Other laboratory tests (eg,, antinuclear antibodies [ANA], anti-Ro/SSA, anti-La/SSB, rheumatoid factor [RF], immunoglobulin levels including immunoglobulin subclasses, HIV testing) are obtained to identify potential causes of secondary lymphoid interstitial pneumonia.
Lung biopsy with demonstration of expansion of the alveolar septae due to lymphocytic and other immune cell (plasma cell, immunoblastic, histiocytic) infiltrates is required for diagnosis in adults. Infiltrates appear occasionally along bronchi and vessels but most commonly along alveolar septa. Immunohistochemical staining and flow cytometry must be done on the tissue to distinguish lymphoid interstitial pneumonia from primary lymphomas. In lymphoid interstitial pneumonia, the infiltrate is polyclonal (both T cells and B cells), whereas other lymphomas produce monoclonal infiltrates. Other common findings include germinal centers and multinucleated giant cells with noncaseating granulomas.
Diagnosis reference
1. Cha SI, Fessler MB, Cool CD, Schwarz MI, Brown KK. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J 2006;28(2):364-369. doi:10.1183/09031936.06.00076705
Treatment of Lymphoid Interstitial Pneumonia
Glucocorticoids or cytotoxic drugs
Treatment of lymphoid interstitial pneumonia, when indicated for symptomatic and/or progressive disease, is with glucocorticoids. Cytotoxic drugs may be added to or used instead of glucocorticoids for patients who have progressive disease despite ongoing treatment, experience adverse effects from glucocorticoids, or require prolonged treatment due to relapsing disease. As with many other causes of interstitial lung diseases, this approach has not been studied in randomized trials and is largely based on prior treatment experience. Treatment is also directed to the cause of lymphoid interstitial pneumonia if present (eg, HIV, immunodeficiency, autoimmunity).
Prognosis for Lymphoid Interstitial Pneumonia
The natural history and prognosis of lymphoid interstitial pneumonia in adults are poorly understood. Spontaneous resolution, resolution after treatment with glucocorticoids or other immunosuppressive medications, progression to lymphoma, or development of pulmonary fibrosis with respiratory insufficiency may ensue. The median survival is estimated to be 11.5 years (1). Common causes of death are infection, development of malignant lymphoma (5%), and progressive fibrosis.
Prognosis reference
1. Cha SI, Fessler MB, Cool CD, Schwarz MI, Brown KK. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J 2006;28(2):364-369. doi:10.1183/09031936.06.00076705
Key Points
Lymphoid interstitial pneumonia is rare.
It is associated with autoimmune disease, immunodeficiencies, and chronic infections (HIV) but can also be idiopathic.
Symptoms and signs tend to be nonspecific but include cough and dyspnea.
Perform a high-resolution CT, bronchoalveolar lavage, and sometimes lung biopsy.
Treat patients with symptomatic or progressive disease with glucocorticoids, cytotoxic drugs, or both; associated disorders need to be treated.







