


Le syndrome de Marfan est une maladie héréditaire rare du tissu conjonctif qui provoque des anomalies oculaires, osseuses, cardiaques, vasculaires, pulmonaires et au niveau du système nerveux central.
Ce syndrome est causé par des mutations du gène codant pour une protéine qui s’appelle fibrilline.
Les symptômes habituels varient de légers à graves et comprennent des bras et des doigts anormalement longs, une hyperlaxité des articulations et des problèmes cardiaques et pulmonaires.
Le diagnostic repose sur les symptômes et les antécédents familiaux.
La plupart des personnes atteintes de ce syndrome atteignent les soixante-dix ans.
Il n’existe aucun traitement curatif du syndrome de Marfan, ni aucune manière de corriger l’altération du tissu conjonctif.
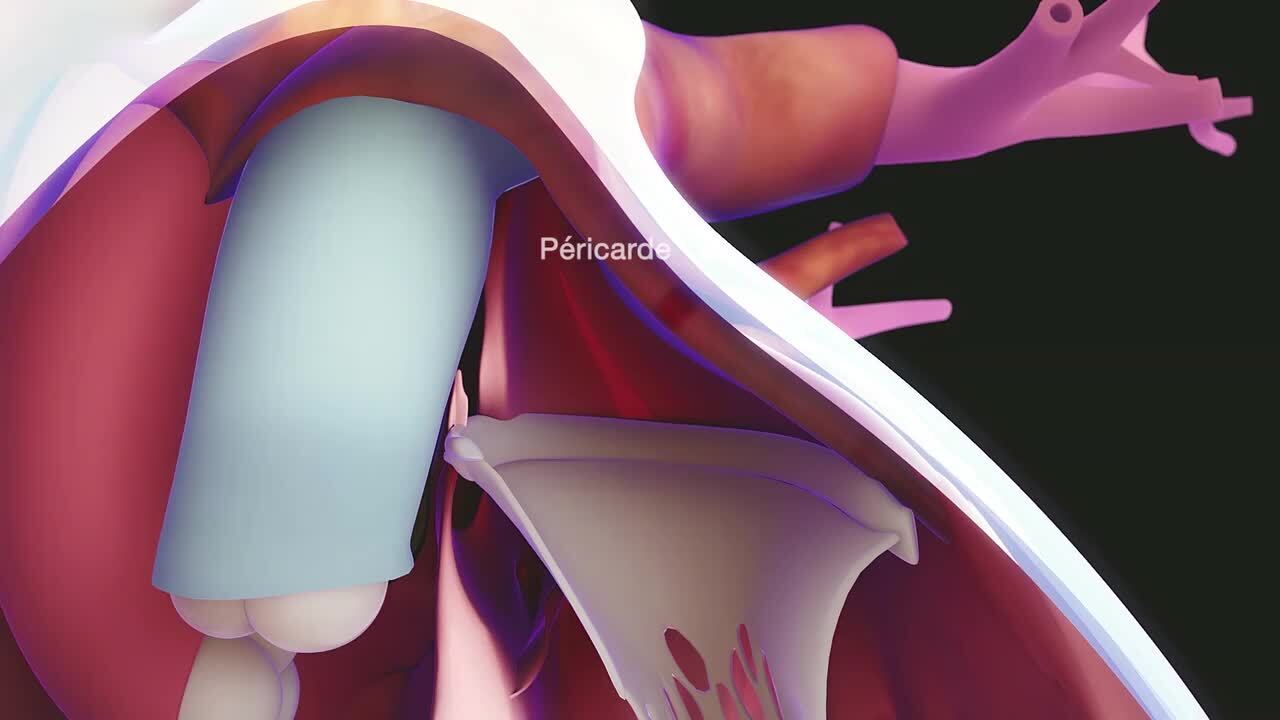
Le syndrome de Marfan est causé par des mutations du gène codant pour une protéine qui s’appelle fibrilline. La fibrilline permet au tissu conjonctif de conserver sa résistance. Le tissu conjonctif est un tissu résistant, souvent fibreux, qui maintient ensemble les différentes structures de l’organisme en leur fournissant un support et de l’élasticité.
Si le gène de la fibrilline est muté, certaines fibres et d’autres parties du tissu conjonctif subissent des changements qui finissent par affaiblir les tissus. Cette fragilisation touche les os, les articulations et des organes internes comme le cœur, les vaisseaux sanguins, les yeux, les poumons et le système nerveux central (cerveau et moelle épinière). Les tissus fragilisés se distendent, se déforment et peuvent se rompre. Par exemple, l’aorte (principale artère de l’organisme) peut être fragilisée, se dilater ou se disséquer. Une fuite d’une valve cardiaque est parfois due à une faiblesse du tissu. Le tissu conjonctif qui réunit habituellement des structures anatomiques peut s’affaiblir ou se rompre, provoquant ainsi la séparation de ces structures. Le cristallin ou la rétine, par exemple, peuvent se détacher de leurs insertions normales.
Symptômes du syndrome de Marfan
Les symptômes du syndrome de Marfan peuvent varier de légers à graves. De nombreuses personnes souffrant du syndrome de Marfan n’en remarquent jamais les symptômes. Chez certains, les symptômes peuvent n’apparaître qu’à l’âge adulte.
Problèmes musculosquelettiques


© Springer Science+Business Media


Photo publiée avec l’aimable autorisation du Dr David D. Sherry.
Les personnes qui souffrent du syndrome de Marfan sont plus grandes que la moyenne des personnes de leur âge ou de leur famille. Elles ont une ouverture alaire (distance entre les pointes des doigts, bras écartés) supérieure à leur taille. Leurs doigts sont longs et fins. Souvent, l’os de thorax (sternum) est déformé en creux ou en relief. Les articulations peuvent être très souples. Les pieds plats, une déformation de l’articulation du genou faisant se courber le genou vers l’arrière, ainsi qu’une courbure anormale du rachis (cyphoscoliose) sont fréquents, tout comme les hernies. Elles ont, en général, un pannicule adipeux sous-cutané peu développé. La voûte palatine est souvent ogivale.
Problèmes cardiaques
Les complications les plus dangereuses sont celles qui affectent le cœur et les poumons. Une faiblesse peut se développer au niveau du tissu conjonctif de la paroi de l’aorte. La paroi fragilisée peut entraîner un suintement sanguin entre les couches internes de la paroi de l’aorte (dissection aortique), ce qui entraîne une déchirure ou une dilatation (anévrisme) susceptible de se rompre. Ces problèmes se développent parfois avant l’âge de 10 ans.
La grossesse augmente le risque de dissection aortique. Pour réduire ce risque, il est souvent recommandé de pratiquer une césarienne.
Si l’aorte s’élargit ou se dilate progressivement, la valve aortique, allant du cœur à l’aorte, peut commencer à fuir (ce que l’on appelle régurgitation aortique). Un élargissement de l’aorte survient chez 50 % des enfants et chez 60 à 80 % des adultes. La valve mitrale, située entre l’oreillette gauche et le ventricule gauche, peut fuir (régurgitation mitrale) ou se prolaber vers l’arrière dans l’oreillette gauche (prolapsus mitral).
Ces altérations valvulaires cardiaques peuvent affecter la capacité du cœur à pomper le sang. Les valves cardiaques altérées peuvent aussi favoriser le développement d’infections graves (endocardite infectieuse).
Problèmes pulmonaires
Des bulles d’air peuvent se former dans les poumons (bulles d’emphysème). Elles peuvent se rompre avec l’entrée d’air dans l’espace entourant le poumon (pneumothorax). Ces troubles peuvent provoquer une douleur et un essoufflement.
Problèmes oculaires
Le cristallin de l’un ou des deux yeux peut se voir affecté (luxé). La personne est très myope (incapable de voir clairement les objets éloignés). La région postérieure de l’œil sensible à la lumière (rétine) peut se décoller du fond de l’œil (voir Décollement de la rétine). La luxation du cristallin et le décollement de la rétine peuvent entraîner une perte définitive de la vision.
Problèmes à la moelle épinière
Le sac entourant la moelle épinière peut s’élargir (ce que l’on appelle ectasie durale). L’ectasie durale est courante chez les personnes atteintes du syndrome de Marfan et survient le plus fréquemment au niveau des parties inférieures du rachis. Elle peut entraîner des céphalées, une lombalgie ou d’autres problèmes neurologiques tels que des fuites urinaires ou fécales.
Diagnostic du syndrome de Marfan
Examen clinique
Analyse génétique
Échocardiographie
Imagerie par résonance magnétique (IRM)
Radiographies
Examens ophtalmologiques
Le médecin peut suspecter un diagnostic de syndrome de Marfan devant une personne inhabituellement grande et maigre, qui, de plus, présente l’un des symptômes caractéristiques du syndrome, ou si le syndrome de Marfan est retrouvé chez d’autres membres de la famille (parents au premier degré tels que le père, la mère, ou un frère ou une sœur). Le médecin base également son diagnostic sur des critères spécifiques concernant la mesure dans laquelle certains systèmes d’organes, tels que le cœur, les yeux et les os, sont affectés.
Une analyse génétique, effectuée en général sur un échantillon de sang, peut aider le médecin à diagnostiquer le syndrome de Marfan.
La surveillance des complications est importante, car elles peuvent entraîner des symptômes sérieux. Chaque année, les patients doivent faire contrôler leur cœur, leurs os et leurs yeux pour voir s’il y a une aggravation. Cet examen annuel inclut généralement une échocardiographie du cœur et de l’aorte, des radiographies des mains, de la colonne vertébrale, du bassin, du thorax, des pieds et du crâne, et un examen ophtalmologique. Une IRM peut également être pratiquée pour évaluer les problèmes cardiaques et cérébraux. À l’apparition de nouveaux symptômes, une échocardiographie et une consultation ophtalmologique sont également réalisées.
Pronostic du syndrome de Marfan
Autrefois, la plupart des personnes qui souffraient de syndrome de Marfan mouraient âgés d’une quarantaine d’années. De nos jours, les personnes atteintes du syndrome de Marfan ont presque la même espérance de vie que les personnes en bonne santé. L’augmentation de l’espérance de vie est probablement due à la prévention de la dissection et de la rupture de l’aorte.
Traitement du syndrome de Marfan
Bêtabloquants
Parfois, réparation chirurgicale de l’aorte et des valves
Parfois un corset, et parfois une réparation chirurgicale, pour la courbure de la colonne vertébrale
Il n’existe aucun traitement curatif du syndrome de Marfan, ni aucune manière de corriger l’altération du tissu conjonctif.
Le traitement du syndrome de Marfan vise à prévenir et/ou à stabiliser les complications potentiellement graves.
Les bêta-bloquants (comme l’aténolol et le propranolol) sont des médicaments qui ralentissent le rythme cardiaque et réduisent la force des contractions cardiaques. Ces médicaments sont administrés pour faire circuler le sang plus doucement dans l’aorte. Toutefois, en présence d’une aorte dilatée ou d’un anévrisme, la partie altérée doit être réparée ou remplacée chirurgicalement. La régurgitation valvulaire sévère est également réparée chirurgicalement. Les femmes enceintes courent un risque particulièrement élevé de complication au niveau de leur aorte, c’est pourquoi une réparation de l’aorte avant la conception doit être envisagée. Des antagonistes des récepteurs de l’angiotensine II (tels que le losartan et le candésartan) sont d’autres médicaments qui peuvent également être administrés pour diminuer la tension artérielle.
Le décollement du cristallin ou de la rétine peut, en général, être traité chirurgicalement.
Un corset est porté aussi longtemps que possible pour traiter toute courbure anormale de la colonne vertébrale (scoliose). Cependant, certains enfants doivent subir une intervention chirurgicale pour corriger cette courbure.
Les personnes doivent recevoir des conseils génétiques. Les personnes et leur famille peuvent obtenir des informations supplémentaires auprès de The Marfan Foundation (Fondation pour le syndrome de Marfan).
Informations supplémentaires
Les ressources en anglais suivantes pourraient vous être utiles. Veuillez noter que LE MANUEL n’est pas responsable du contenu de cette ressource.
The Marfan Foundation (Fondation pour le syndrome de Marfan) : Une ressource qui apporte un soutien, du contenu éducatif et des informations communautaires sur le syndrome de Marfan





