



- Загальні відомості про захворювання ниркових клубочків
- Загальні відомості про нефритичний синдром
- Синдром Альпорта
- Нефропатія імуноглобуліну А
- Постінфекційний гломерулонефрит (ПІГН)
- Швидкопрогресуючий гломерулонефрит (ШПГН)
- Хвороба тонких базальних мембран
- Загальні відомості про нефротичний синдром
- Вроджені нефротичні синдроми
- Діабетична нефропатія
- Вогнищевий сегментарний гломерулосклероз
- ВІЛ-асоційована нефропатія
- Мембранозна нефропатія
- Хвороба мінімальних змін
- Фібрилярні та імунотактоїдні гломерулопатії
- Вовчаковий нефрит
- Мембранопроліферативний гломерулонефрит
Membranoproliferative glomerulonephritis (MPGN) is characterized by a pattern of glomerular injury on light microscopy, including hypercellularity and thickening of the glomerular basement membrane. The clinical presentation usually consists of mixed nephritic and nephrotic features. Cause is idiopathic or secondary to another disorder. Diagnosis is by renal biopsy. Treatment is directed at the underlying disorder, when present. For patients with idiopathic disease, treatment may be supportive or include corticosteroids and other immunosuppressive agents.





(See also Overview of Nephrotic Syndrome.)
Membranoproliferative glomerulonephritis (MPGN) is characterized histologically by glomerular basement membrane (GBM) thickening and proliferative changes on light microscopy. Historically, MPGN was classified into types I, II, and III, based on the location of deposits on electron microscopy examination:
Type I: Characterized by electron-dense deposits in the mesangium and subendothelial space, consisting of both immunoglobulin and C3.
Type II (dense deposit disease): Characterized by electron-dense ribbon-like deposits along the basement membranes of the glomeruli, tubules, and Bowman's capsule, consisting mostly of complement.
Type III: Characterized by both subepithelial and subendothelial deposits.
However, advances in the understanding of the pathogenesis of MPGN has revealed that the classification based on electron microscopy has limitations and can result in overlap among the different types.
The preferred classification is based on pathophysiologic processes and informed by findings on immunofluorescence microscopy. This classification scheme broadly divides MPGN into
Immunoglobulin/immune complex–mediated MPGN
Complement-mediated MPGN
MPGN without immunoglobulin or complement deposition
An underlying (secondary) cause is present in the majority of cases regardless of type. Idiopathic (primary) cases are less common and usually observed within the immunoglobulin/immune complex–mediated category.
Primary forms affect children and young adults between ages 8 and 30 and account for 10% of cases of nephrotic syndrome in children; secondary forms tend to affect adults > 30. Men and women are affected equally. Reported familial cases of some types suggest genetic factors play a role in at least some cases. Many factors contribute to hypocomplementemia.

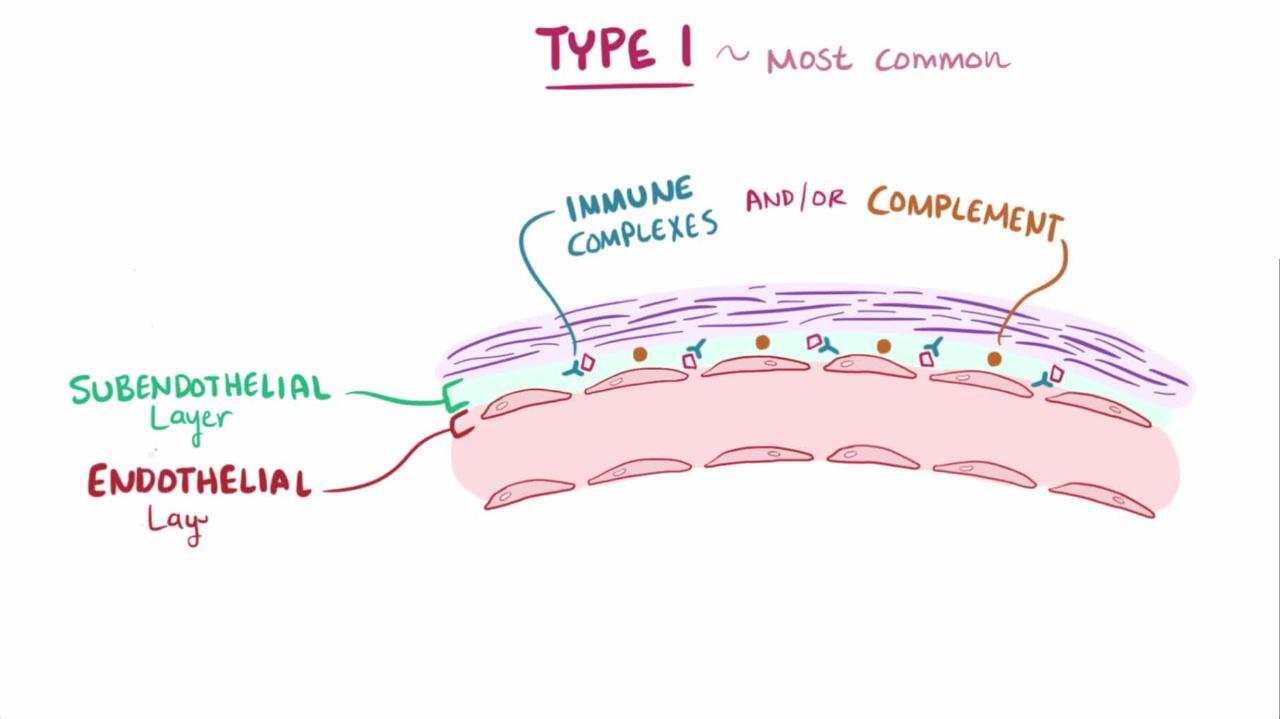
МПГН, опосередкований імуноглобулінами/імунними комплексами
Immunoglobulin/immune complex–mediated MPGN is due to chronic antigenemia and/or circulating immune complexes and most commonly occurs secondary to one of the following:
Systemic immune complex disorder (eg, systemic lupus erythematosus, mixed connective tissue disease, Sjögren syndrome, rheumatoid arthritis)
Chronic infection (eg, bacterial endocarditis, HIV infection, hepatitis B or C infection, visceral abscess, ventriculoatrial shunt infection, protozoa infection)
Monoclonal gammopathies (eg, monoclonal gammopathy of unknown significant (MGUS), chronic lymphocytic leukemia, lymphomas, multiple myeloma)
Immunofluorescence microscopy findings may suggest certain underlying diseases.


Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
МПГН, опосередкований комплементом


Image provided by Agnes Fogo, MD, and the American Journal of Kidney Diseases' Atlas of Renal Pathology (see www.ajkd.org).
Complement-mediated MPGN is less common than immunuglobulin/immune complex–mediated MPGN. It is caused by dysregulation and persistent activation of the alternative complement pathway, and there is deposition of complement products along the capillary walls and in the mesangium. Complement-mediated MPGN can be further categorized based on features observed on immunofluorescence and electron microscopy as C3 or C4 glomerulonephritis or dense deposition disease (DDD).
Мембранопроліферативний тип МПГН без відкладення імуноглобуліну або комплементу
MPGN without immunoglobulin or complement deposition may be seen in:
Healing phase of thrombotic microangiopathies (eg, thrombotic thrombocytopenia purpura,hemolytic uremic syndrome)
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin changes)
Radiation nephritis
Nephropathy associated with bone marrow transplantation
Drug-associated thrombotic microangiopathies
Disfibrinogenemia and other prothrombotic states
Histology suggests endothelial injury and reparative changes. Immunofluorescence microscopy does not show significant immunoglobulin or complement deposition, and electron microscopy does not show electron-dense deposits along the capillary walls.
Symptoms and Signs of Membranoproliferative Glomerulonephritis
Symptoms and signs are similar to those for other types of glomerulonephritis. The urine sediment may reveal hematuria (with dysmorphic red cells and/or red cell casts). The degree of proteinuria is variable. The serum creatinine may be normal or elevated.
Patients with dense deposition disease, a subtype of complement-mediated MPGN, have a greater incidence of ocular abnormalities (basal laminar drusen, diffuse retinal pigment alterations, diskiform macular detachment, choroidal neovascularization), which ultimately impair vision.
Diagnosis of Membranoproliferative Glomerulonephritis
Renal biopsy
Serum complement profile
Specific laboratory tests based on the classification of the membranoproliferative lesion and associated underlying disorders
Diagnosis is confirmed by renal biopsy. The pattern of immunoglobulin and complement deposition on immunofluorescence microscopy helps classify the type of MPGN lesion. Additional tests are also done to help identify the underlying cause of the MPGN lesion.
Hypocomplementemia is more frequently present in all types of MPGN than in other glomerular disorders and provides supportive evidence of the diagnosis (see table Serum Complement Profiles in Membranoproliferative Glomerulonephritis). In immunoglobulin/immune complex–mediated MPGN, the classic complement pathway is activated; C3 is normal or mildly decreased, and C4 is typically decreased. In complement-mediated MPGN, the alternate complement pathway is activated; C3 is decreased, but C4 is normal. In MPGN without immunoglobulin complement deposition, C3 and C4 are normal.
Профілі сироваткового комплементу при Мембранопроліферативному гломерулонефриті
Classification Based on Immunofluorescence Microscopy | Complement Profile |
|---|---|
Immunoglobulin/immune complex–mediated MPGN | Classic complement pathway activated C3: Normal or mildly decreased C4: Decreased |
Complement-mediated MPGN | Alternate complement pathway activated C3: Decreased C4: Normal |
MPGN without immunoglobulin or complement deposition | C3: Normal C4: Normal |
Specific laboratory testing for secondary causes is based on their associations with the types of MPGN.
Complete blood count (CBC), often obtained in the course of diagnostic evaluation, demonstrates normochromic-normocytic anemia, often out of proportion to the stage of renal insufficiency (possibly because of hemolysis), and thrombocytopenia from platelet consumption.
Treatment of Membranoproliferative Glomerulonephritis
Corticosteroids for children with nephrotic-range proteinuria
Kidney transplantation for patients with end-stage kidney disease
Underlying disorders are treated when possible. Patients with mild disease (ie, normal kidney function and non–nephrotic-range proteinuria or significant hematuria) are typically treated with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs). Additional immunosuppressive therapy is typically not indicated in such patients.
Among patients with nephrotic-range proteinuria and normal renal function, treatment with corticosteroids (eg, prednisone 2.5 mg/kg orally once a day on alternate days [maximum 80 mg/day]) for 1 year, followed by tapering to maintenance therapy may stabilize renal function. However, corticosteroid treatment may retard growth in children and cause hypertension. Calcineurin inhibitors (eg, cyclosporine or tacrolimus) may be used as alternatives for patients who do not tolerate or wish to receive corticosteroids. Patients who present with rapidly progressive crescentic MPGN should be treated more aggressively with corticosteroids plus cyclophosphamide.
Other general supportive measures include dietary and sodium restriction, antihypertensive therapy, renin-angiotensin inhibition, and treatment of dyslipidemia.
Prognosis for Membranoproliferative Glomerulonephritis
Prognosis is good if a condition causing secondary membranoproliferative glomerulonephritis is successfully treated. As with other glomerular disorders, the outcome tends to be worse if patients present with proteinuria in the nephrotic range.
Додаткова інформація
The following English-language resource may be useful. Please note that THE MANUAL is not responsible for the content of this resource.
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group: KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 100(4S):S1-S276, 2021. doi: 10.1016/j.kint.2021.05.021
Ключові моменти
Membranoproliferative glomerulonephritis (MPGN) is a pattern of glomerular injury with characteristic findings on light microscopy, including hypercellularity and thickening of the glomerular basement membrane.
Patients most often present with nephrotic syndrome, but they may present with nephritic syndrome.
Confirm the diagnosis with renal biopsy and obtain serum complement (C3, C4) and additional tests to classify MPGN as immune complex/monoclonal immunoglobulin-mediated MPGN, complement-mediated MPGN, or MPGN without immunoglobulin or complement deposition.
Test for underlying diseases based on clinical presentation and specific type of MPGN.
Treat nephrotic range proteinuria with corticosteroids and consider immunosuppressants depending upon progression of renal dysfunction; target additional therapy at any specific underlying disease.


