




A biodisponibilidade refere-se à extensão e à velocidade em que a porção ativa (fármaco ou metabólito) adentra a circulação sistêmica, alcançando, assim, o local de ação.
A biodisponibilidade de um fármaco é predominantemente determinada pelas propriedades da forma de dosagem, que dependem, em parte, de sua forma e fabricação. Diferenças na biodisponibilidade entre formulações de um determinado fármaco podem ter significado clínico; assim, é essencial saber se as formulações medicamentosas são equivalentes.
A equivalência química indica que as formulações possuem os mesmos compostos ativos e quantidades, além de obedecerem aos padrões oficiais atualizados, mas pode haver diferenças quanto aos ingredientes inativos nas diferentes formulações do fármaco. A bioequivalência indica que as formulações, quando administradas ao mesmo paciente e no mesmo esquema de dosagem, resultam em concentrações equivalentes do fármaco no plasma e nos tecidos. A equivalência terapêutica indica que as formulações, quando administradas para o mesmo paciente e no mesmo esquema de dosagem, têm efeitos terapêuticos e adversos iguais.
Espera-se que os produtos bioequivalentes sejam terapeuticamente equivalentes. A ausência de equivalência terapêutica (p. ex., mais efeitos adversos e menos eficácia) é geralmente identificada durante o tratamento de longo prazo, quando se administra um substituto não equivalente a pacientes que se encontram estabilizados com uma determinada formulação.
Às vezes, a equivalência terapêutica é possível, apesar de diferenças na biodisponibilidade. Por exemplo, o índice terapêutico (razão da concentração tóxica mínima para a concentração efetiva média) da penicilina é tão amplo que a eficácia e a segurança geralmente não são comprometidas pelas diferenças moderadas na concentração plasmática decorrentes das desigualdades na biodisponibilidade dos produtos da penicilina. Em contrapartida, para os fármacos com índice terapêutico relativamente estreito, as diferenças na biodisponibilidade podem causar falta substancial de equivalência terapêutica.
(Ver também Visão geral da farmacocinética.)
Causas da baixa biodisponibilidade
Fármacos administrados por via oral devem passar pela parede intestinal e, em seguida, pela circulação portal até o fígado; ambos são locais comuns de metabolismo de primeira passagem (metabolismo que ocorre antes de um fármaco alcançar a circulação sistêmica). Assim, muitos fármacos podem ser biotransformados antes de alcançar concentrações plasmáticas adequadas. A biodisponibilidade baixa é mais comum com as formas de dosagem oral de fármacos com absorção lenta e baixa hidrossolubilidade.
O tempo insuficiente de absorção no trato gastrointestinal é causa comum de biodisponibilidade baixa. Se o fármaco não se dissolver rapidamente ou não conseguir penetrar na membrana epitelial (p. ex., se for intensamente ionizada ou polar), o tempo no local de absorção pode ser insuficiente. Nesses casos, a biodisponibilidade tende a ser amplamente variável, bem como baixa.
Idade, sexo, atividade física, fenótipo genético, estresse, doenças (p. ex., acloridria e síndromes de má absorção) ou cirurgia pregressa do trato gastrointestinal (p. ex., cirurgia bariátrica) também podem afetar a biodisponibilidade de um fármaco.
Reações químicas que reduzem a absorção podem diminuir a biodisponibilidade. Elas incluem a formação de um complexo (p. ex., entre tetraciclina e íons metálicos polivalentes), hidrólise por ácido gástrico ou enzimas digestivas (p. ex., penicilina e hidrólise do palmitato de cloranfenicol), conjugação na parede intestinal (sulfoconjugação de isoproterenol), adsorção de outros fármacos (p. ex., de digoxina para colestiramina) e metabolismo por flora do lúmen.
Avaliação da biodisponibilidade
A biodisponibilidade costuma ser avaliada determinando-se a área sob a curva de concentração plasmática em relação ao tempo (ver figura Relação representativa entre concentração plasmática e tempo após uma dose oral única de um fármaco hipotético). A medida mais confiável da biodisponibilidade de um fármaco é AUC. A AUC é diretamente proporcional à quantidade total de fármaco inalterado que alcança a circulação sistêmica. Os produtos dos fármacos podem ser considerados bioequivalentes quanto à extensão e velocidade de absorção se suas curvas de concentração plasmática forem essencialmente superimpostas.
Relação representativa entre concentração plasmática e tempo após uma dose oral única de um fármaco hipotético

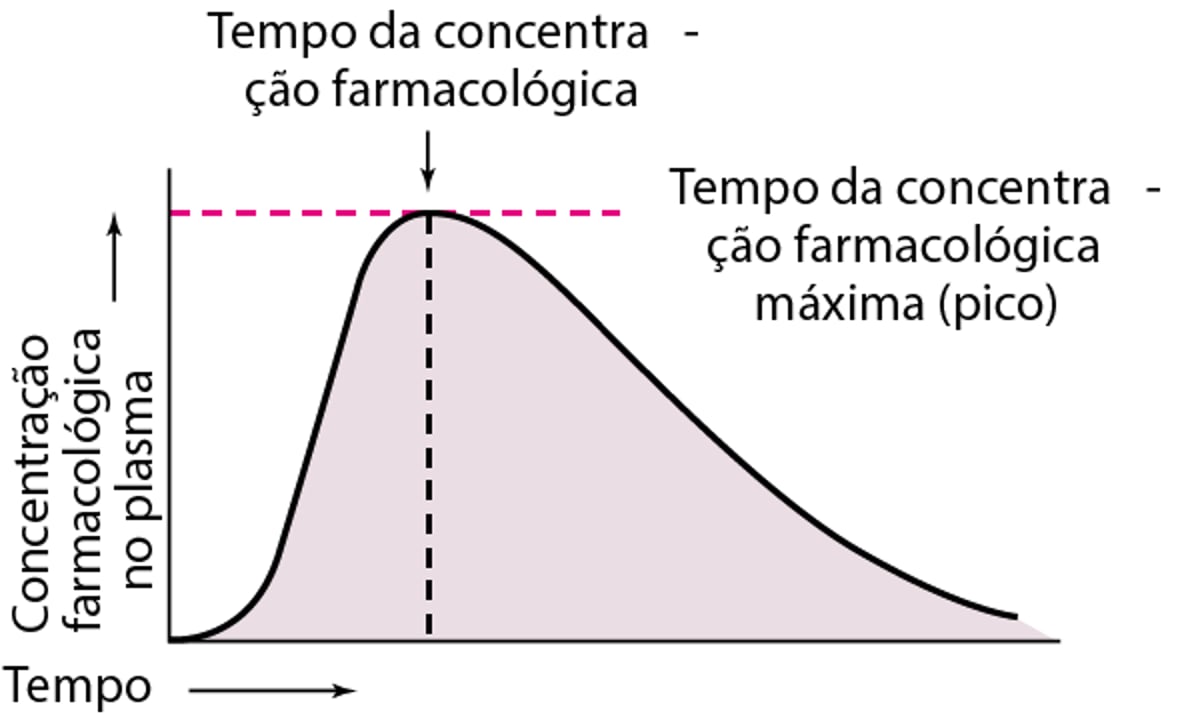
A concentração plasmática do fármaco aumenta com a extensão da absorção; a máxima (pico) é alcançada quando a taxa de eliminação do fármaco iguala-se à taxa de absorção. As determinações de biodisponibilidade com base na concentração plasmática máxima podem ser errôneas, porque a eliminação do fármaco inicia-se assim que este entra na corrente sanguínea. O tempo de pico (quando o fármaco atinge concentração plasmática máxima) constitui o índice geral mais utilizado para a avaliação da absorção; quanto mais lenta a absorção, mais tardio é o tempo de pico.
Para os fármacos excretados primariamente inalterados na urina, pode-se estimar a biodisponibilidade pela avaliação da quantidade total de fármaco excretado após dose única. Idealmente, coleta-se a urina por um período de 7 a 10 meias-vidas de eliminação para a recuperação urinária completa do fármaco absorvido. Após múltiplas doses, estima-se a biodisponibilidade pela avaliação do fármaco inalterado recuperado na urina durante o período de 24 horas sob condições estáveis.










