




遺伝性腎炎は,遺伝的に雑多な疾患であり,腎炎症候群(例,血尿,タンパク尿,高血圧,最終的に腎機能不全)を特徴とし,しばしば感音難聴を伴い,より頻度は低いが眼症状が発生する。原因は,IV型コラーゲンに影響を及ぼす遺伝子の突然変異である。診断は家族歴などの病歴,尿検査,生検(腎または皮膚)による。治療は慢性腎臓病の治療と同じであり,ときに腎移植が含まれる。
(腎炎症候群の概要も参照のこと。)
遺伝性腎炎は,IV型コラーゲンα5鎖をコードするCOL4A3,COL4A4,およびCOL4A5遺伝子の突然変異,およびその結果もたらされる変性IV型コラーゲン鎖に起因する腎炎症候群である。コラーゲン変性が糸球体疾患をもたらす機序は不明であるが,組織と機能の障害が推定され,大部分の家系で糸球体と尿細管基底膜の肥厚と菲薄化が起こり,巣状あるいは局所的な基底板の多層化(網目状パターン)がみられる。糸球体瘢痕および間質線維化が最終的にもたらされる。

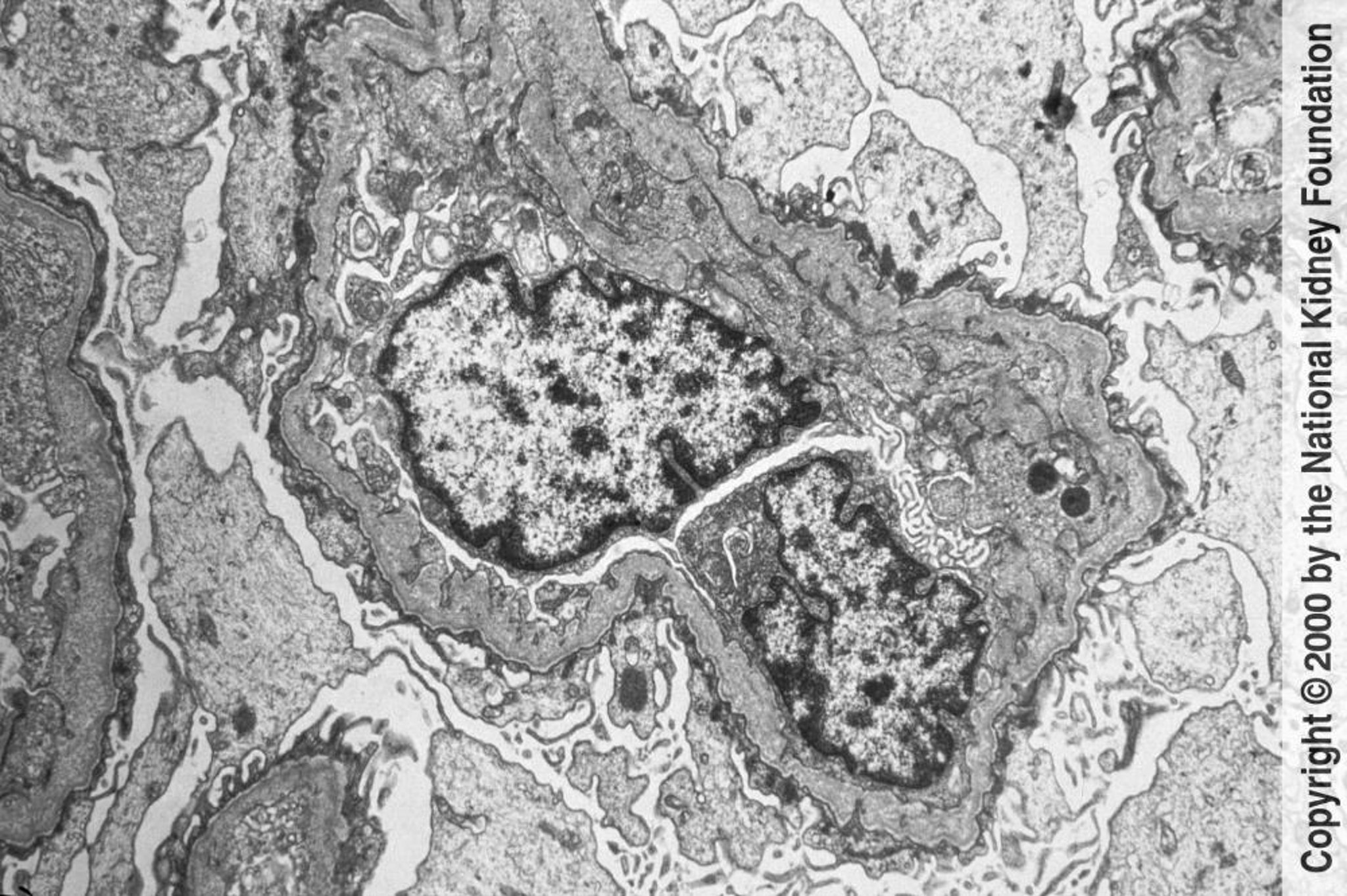
Image provided by Agnes Fogo, MD, and the American Journal of Kidney Disease’s Atlas of Renal Pathology (see www.ajkd.org).
本症は最も一般的にはX連鎖の形式で遺伝するが,常染色体劣性およびまれに常染色体優性の形式で遺伝するものも存在する。X連鎖遺伝の症例は,臨床的に以下のように分類されることがある:
若年型:腎機能不全が20~30歳の間に発生する
成人型:腎機能不全が30歳以降に発生する
症状と徴候
男性における古典的なX連鎖型と常染色体劣性型は臨床的に類似する。患者は,最終的に急性腎炎症候群に類似した腎の症状と徴候を発症し(例,顕微鏡的血尿,高血圧,最終的にタンパク尿を伴う肉眼的血尿),腎機能不全に20歳から30歳の間に進行する(若年型)。
高周波音域に影響する感音難聴がしばしば存在し,幼児期には認識されない場合がある。
眼科的異常として白内障(最も頻度が高い),前部円錐水晶体(水晶体包の菲薄化により水晶体の前面での規則的な円錐状の突出),球状水晶体(球状の水晶体変形で,水晶体亜脱臼の素因の可能性),眼振,網膜色素変性,失明も発生するが,難聴と比較して発生頻度は低い。
X連鎖型はヘテロ接合の女性に発生し,その場合正常なX染色体が1つあるため,男性と比べて通常は症状がより軽度で進行が遅い。
X連鎖型の男性患者の一部は,30歳以降に腎機能不全を発症し,難聴は後期に発生するか軽度であり,常染色体優性型では一般的に45歳以降まで腎不全は生じない(成人型)。
X連鎖型のアルポート症候群患者では,感音難聴は通常小児期に生じるが,腎疾患は小児期には生じないことが多い。
診断
尿検査
腎生検
分子遺伝学的検査
診断は,尿検査で顕微鏡的血尿を呈するか,肉眼的血尿の再発性エピソードを呈し,特に聴覚もしくは視覚の異常または慢性腎臓病の家族歴を有する患者で示唆される。
尿検査および通常は腎生検を施行する。また,尿中には変形赤血球に加え,タンパク質,白血球,様々な種類の円柱が認められる場合がある。ネフローゼ症候群がまれに起こる。光学顕微鏡検査では,特徴的な組織学的変化は認められない。診断は以下のいずれかによって確定することができる:
腎生検とIV型コラーゲンサブタイプに対する免疫染色法の併用
糸球体毛細血管基底膜の多様な肥厚および菲薄化を伴う基底板の特徴的な組織崩壊が電子顕微鏡を用いて確認される
家族歴が陽性の患者に対する皮膚生検とIV型コラーゲンサブタイプに対する免疫染色法の併用
COL4A遺伝子の分子遺伝学的検査
遺伝性腎炎を一部の病型の菲薄基底膜病と鑑別するためには,免疫染色と電子顕微鏡観察の併用がしばしば必要となる。
治療
要点
血尿と聴覚および/または視覚異常がみられる患者と慢性腎臓病の家族歴がある患者では,遺伝性腎炎を考慮する。
診断は,腎またはときに皮膚の生検およびIV型コラーゲンサブタイプの免疫染色法または分子遺伝学的検査により確定する。
慢性腎臓病を治療し,移植を考慮する。








