


Granulomatosis with polyangiitis is characterized by necrotizing granulomatous inflammation, small- and medium-sized vessel vasculitis, and focal necrotizing glomerulonephritis, often with crescent formation. Typically, the upper and lower respiratory tract and the kidneys are affected, but any organ may be. Symptoms vary depending on the organs and systems affected. Patients may present with upper and lower respiratory tract symptoms (eg, recurrent nasal discharge or epistaxis, cough), followed by hypertension and edema, or with symptoms reflecting multiorgan involvement. Diagnosis usually requires biopsy. Treatment is with corticosteroids plus another immunosuppressant. Remission is usually possible, although relapses are common.
(See also Overview of Vasculitis.)
The incidence of granulomatosis with polyangiitis (GPA) varies depending on the population being studied. For example, a large cohort study in the United Kingdom reported an incidence of 11.8/million person-years (1). It is most common among people of European ancestry but can occur in all ethnic groups and at any age (2). Mean age at onset is 40 years.
The cause of GPA is unknown, although immunologic mechanisms play a role. Most patients with active generalized disease have antineutrophil cytoplasmic antibodies (ANCA).
General references
1. Pearce FA, Grainge MJ, Lanyon PC, Watts RA, Hubbard RB. The incidence, prevalence and mortality of granulomatosis with polyangiitis in the UK Clinical Practice Research Datalink. Rheumatology (Oxford) 56(4):589-596, 2017. doi:10.1093/rheumatology/kew413
2. Terrier B, Dechartres A, Deligny C, et al. Granulomatosis with polyangiitis according to geographic origin and ethnicity: clinical-biological presentation and outcome in a French population. Rheumatology (Oxford) 56(3):445-450, 2017. doi:10.1093/rheumatology/kew423
Pathophysiology of Granulomatosis with Polyangiitis
Characteristically, granulomas form with histiocytic epithelioid cells and often with multinucleated giant cells. Plasma cells, lymphocytes, neutrophils, and eosinophils are also present. Inflammation affects tissues as well as vessels; vasculitis may be a small or large component of the disease. Micronecrosis, usually with neutrophils (microabscesses), occurs early. Micronecrosis progresses to macronecrosis. A central area of necrosis (called geographic necrosis) is rimmed by lymphocytes, plasma cells, macrophages, and multinucleated giant cells. A zone of fibroblastic proliferation with palisading histiocytes may surround the area.
Nonspecific chronic inflammation and tissue necrosis occur in the nose. The lungs are most likely to display the full spectrum of histopathologic abnormalities, but diagnostic features are not typically identified on the small tissue samples obtained by transbronchial biopsy. In the kidneys, the most common finding is a pauci-immune crescentic focal glomerulonephritis with necrosis and thrombosis of individual loops or larger segments of the glomerulus. Vasculitic lesions and disseminated granulomas occur only occasionally.
Symptoms and Signs of Granulomatosis with Polyangiitis
Onset of granulomatosis with polyangiitis may be insidious or acute; the full spectrum of the disease may take years to evolve. Some patients present initially with upper and lower respiratory tract symptoms; at some point later, the kidneys are affected. In other patients, onset of systemic manifestations is relatively acute; several organs and systems, such as the upper respiratory tract, peripheral nervous system (causing multiple mononeuropathy [mononeuritis multiplex]), kidneys (causing glomerulonephritis), and lower respiratory tract (causing hemorrhage, lung nodules, cavities, or a combination), are simultaneously affected.
Upper respiratory tract: Sinus pain, serosanguineous or purulent discharge, and epistaxis may occur. The mucosa appears granular (like cobblestones) and is friable; ulcers, thick dark crusts, and septal perforation are common. Nasal chondritis can occur with swelling, pain, and collapse of the nasal bridge (saddle nose). Patients may report recurrent sinusitis that has responded inadequately to multiple antibiotic regimens and has required one or more sinus operations before diagnosis. Secondary infections (eg, due to Staphylococcus aureus) may develop. Subglottic stenosis may develop, causing symptoms such as pain in the larynx, hoarseness, dyspnea, wheezing, and stridor.
Ears: Otitis, sensorineural hearing loss, vertigo, and chondritis may occur. The middle ear, inner ear, and mastoids are often affected.
Eyes: Eyes may appear red and swollen. Nasolacrimal duct inflammation and obstruction, conjunctivitis, scleritis, uveitis, or retinal vasculitis may also occur. Inflammatory infiltrates in the retro-orbital space (orbital pseudotumor) can cause proptosis, compression of the optic nerve, and blindness. Extension into the extraocular muscles leads to diplopia. If serious eye symptoms develop, evaluation and treatment are required immediately to prevent permanent vision loss.
Lower respiratory tract: Respiratory manifestations are common. Inflammation of the major bronchi and branches can cause localized wheezing, postobstructive pneumonia, and atelectasis. Single or multiple pulmonary nodules, with or without cavitation, and parenchymal infiltrates, sometimes cause symptoms, such as chest pain, shortness of breath, and productive cough. Dyspnea with bilateral infiltrates, with or without hemoptysis, may indicate alveolar hemorrhage and must be evaluated immediately.
Heart: Coronary artery disease may occur but rarely.
Musculoskeletal system: Patients frequently present with myalgias, arthralgias, or nonerosive inflammatory arthritis.
Skin: Palpable purpura, tender subcutaneous nodules, papules, livedo reticularis, or ulcers may develop.
Nervous system: Vasculitis may cause ischemic peripheral neuropathy, brain lesions, or extension of inflammation into neural tissue from contiguous sites. Lesions that originate in the sinuses or middle ear may extend directly to the retropharyngeal area and base of the skull, leading to cranial neuropathy, proptosis, argininevasopressin deficiency, or meningitis.
Kidneys: Symptoms and signs of glomerulonephritis develop. Urinary sediment is frequently abnormal, and serum creatinine may increase rapidly. Edema and hypertension may result. Rapidly progressive glomerulonephritis, which is life threatening, can develop.
Venous system: Deep venous thrombosis can affect the lower extremities mostly when granulomatosis with polyangiitis is active.
Other organs: Occasionally, an inflammatory mass occurs in the breasts, kidneys, prostate, or other organs.

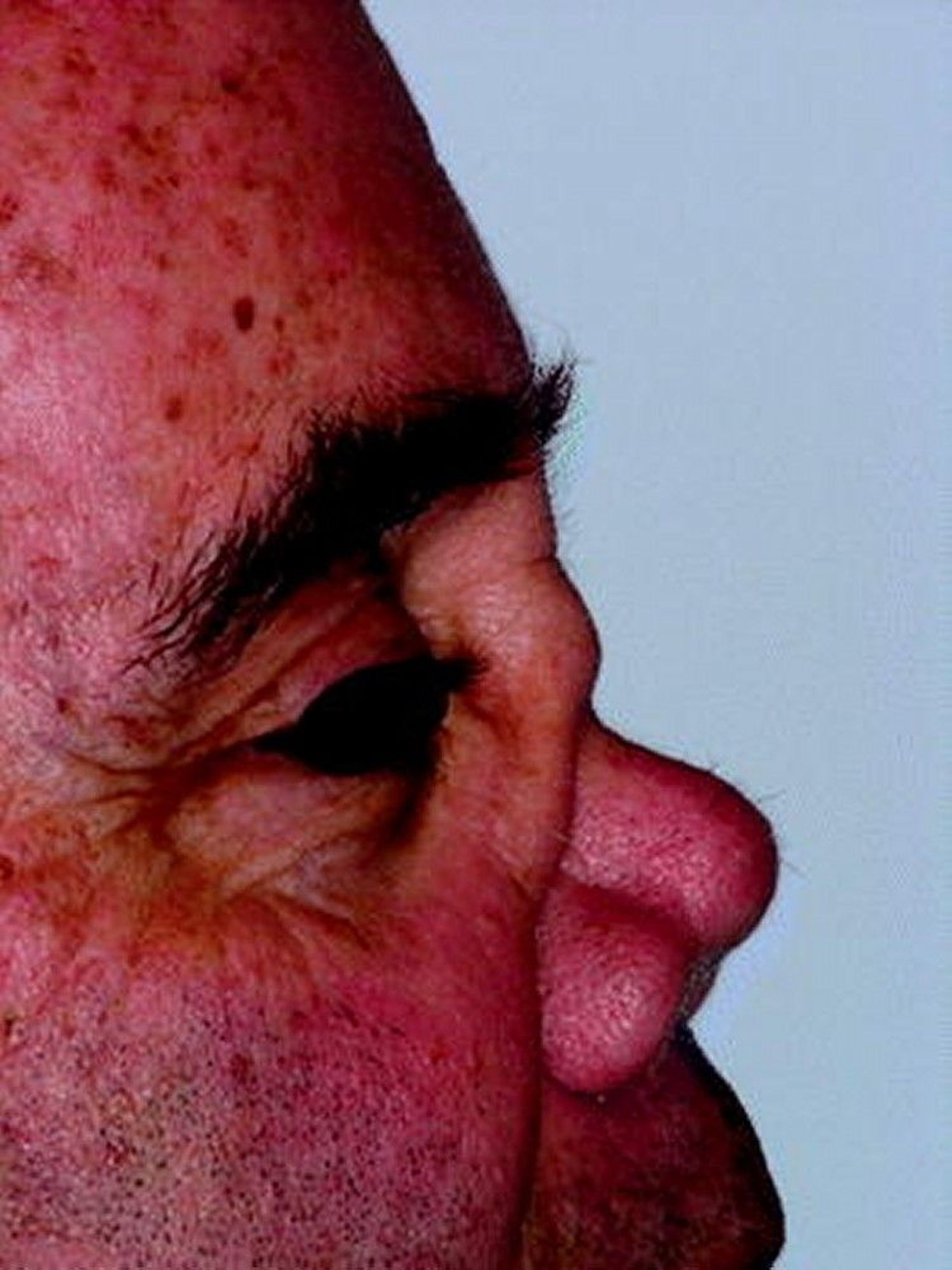
© Springer Science+Business Media

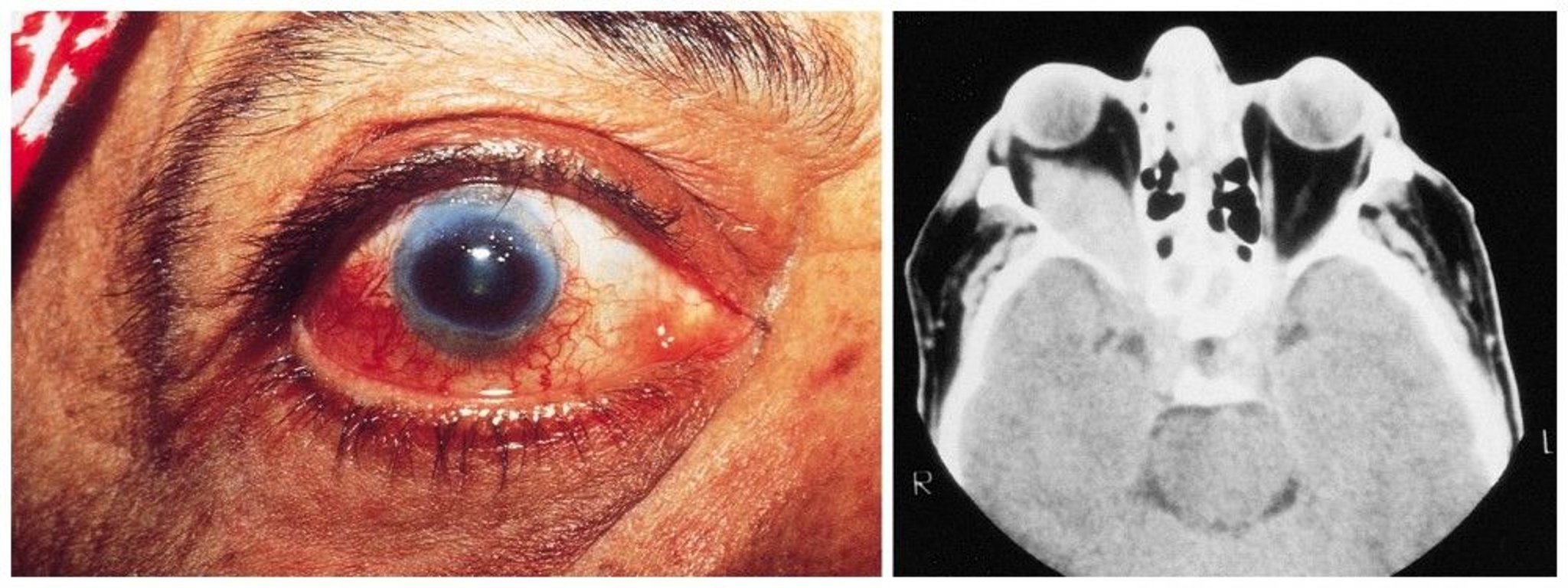
© Springer Science+Business Media
Diagnosis of Granulomatosis with Polyangiitis
Routine laboratory tests, including urinalysis
Tests for antineutrophil cytoplasmic antibodies
Chest and sinonasal CT
Biopsy for definitive diagnosis
Granulomatosis with polyangiitis should be suspected in patients with chronic, unexplained respiratory symptoms and signs (including otitis media in adults), particularly if manifestations in other organ systems, especially the kidneys, also suggest the disorder. Routine laboratory tests are done, but ANCA testing and selective biopsy of involved tissues yield the most specific findings. Biopsy of nasal tissue only rarely provides a definite diagnosis.
Routine laboratory tests include erythrocyte sedimentation rate, C-reactive protein, complete blood count with differential, serum albumin and total protein, serum creatinine, urinalysis, 24-hour urine protein, and chest radiograph. Sinonasal CT may show sinus mucosal thickening or opacification, nasal septal perforation, and bone damage. Chest CT without contrast should always be performed in patients with suspected disease because the chest radiograph may miss nodules, masses, and/or cavitary lesions. In most patients with active disease, erythrocyte sedimentation rate and C-reactive protein are elevated, and serum albumin and total protein are decreased; anemia and thrombocytosis are sometimes detected. Dysmorphic red blood cells and red blood cell casts, detected during urinalysis, indicate glomerular involvement. Proteinuria may be detected. Serum creatinine may be increased.

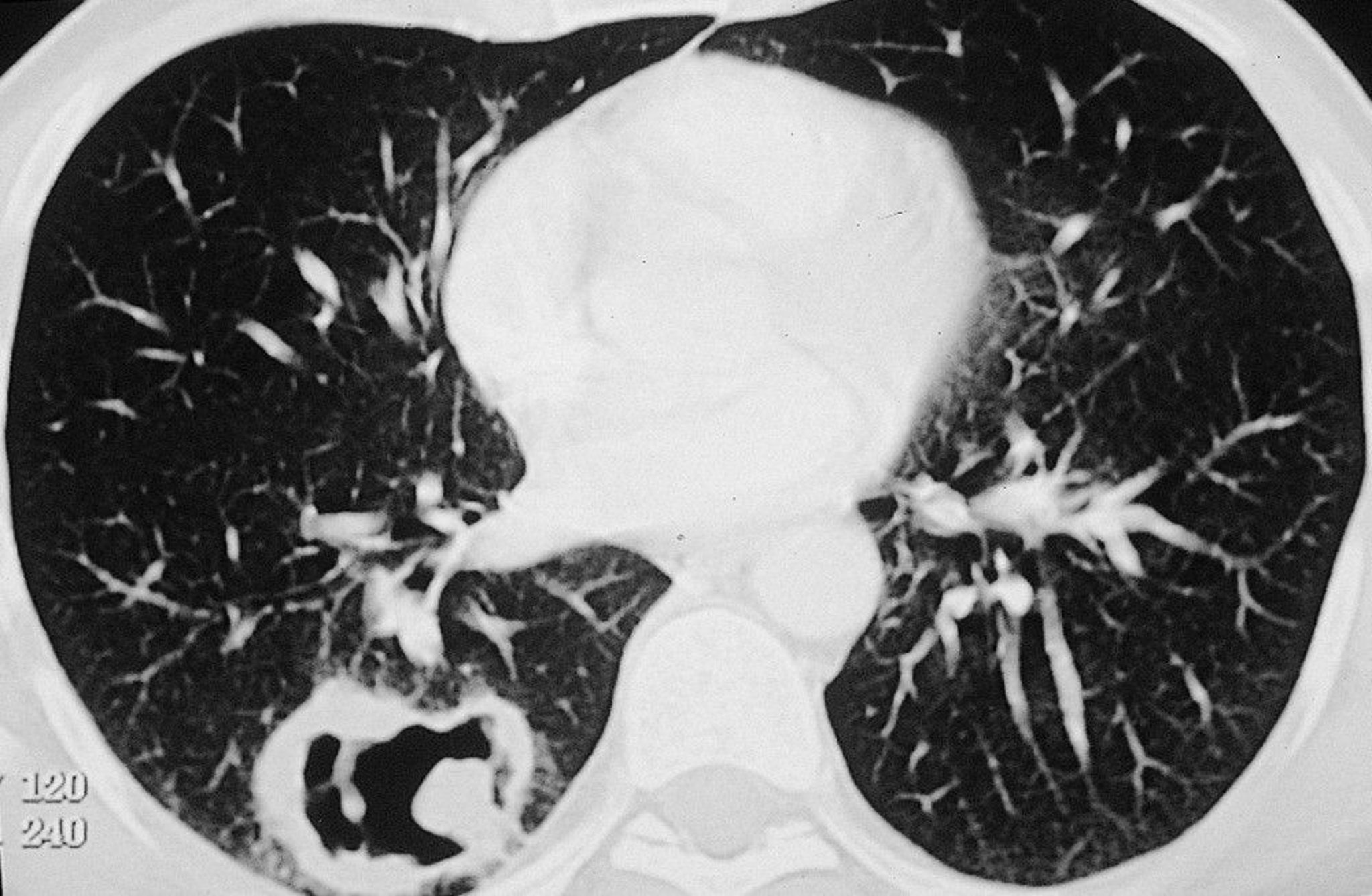
© Springer Science+Business Media
Serologic testing to detect antineutrophil cytoplasmic antibodies (ANCA) is followed by enzyme-linked immunosorbent assay (ELISA) to check for specific antibodies. Most patients with active disease have cytoplasmic ANCA (c-ANCA), with antibodies against proteinase-3 (PR3); these findings plus characteristic clinical findings suggest GPA.
Some patients with other disorders (eg, bacterial endocarditis, cocaine use disorder, systemic lupus erythematosus, amebiasis, tuberculosis) test positive for ANCA. Because tests for rare diseases are more likely to be falsely positive when ordered for patients without a high pretest probability for the disease and the positive predictive value of a positive ANCA test is around 50% (1), ANCA testing should be reserved for patients in whom the pretest probability for GPA or another ANCA-associated vasculitis is at least moderately high (eg, patients with alveolar hemorrhage, glomerulonephritis, or multiple mononeuropathy plus other features of microscopic polyangiitis or GPA) (2).
A positive ANCA test does not exclude mycobacterial and fungal infections; thus, patients with positive ANCA results and cavitary lung lesions still require bronchoscopy and adequate cultures and other tests for tuberculosis and fungal infections. ANCA testing (titre) should not be used to guide subsequent treatment. During apparent remission, ANCA may increase or ANCA test results may change from negative to positive. In some of these patients, symptoms do not recur; in others, symptoms recur or worsen soon after the test is done or during the next few weeks, months, or sometimes years.
Biopsy should be done if possible to confirm the diagnosis of GPA. Clinically abnormal sites may be biopsied first. Biopsy of affected lung tissue is most likely to reveal characteristic findings; open thoracotomy provides the best access. Biopsies of lung or sinus tissue are cultured to exclude infection. Renal biopsy that shows pauci-immune necrotizing focal crescentic or noncrescentic glomerulonephritis strongly supports the diagnosis. Biopsy results of various tissues may also provide histologic information that can help guide treatment (eg, renal fibrosis).
Differential diagnosis includes other vasculitic disorders that affect small- and medium-sized vessels. Infections, especially those due to slow-growing fungi or acid-fast organisms, should be excluded by staining and culture of the sampled tissues.
Diagnosis references
1. McLaren JS, Stimson RH, McRorie ER, Coia JE, Luqmani RA. The diagnostic value of anti-neutrophil cytoplasmic antibody testing in a routine clinical setting. QJM 94(11):615-621, 2001. doi:10.1093/qjmed/94.11.615
2. Guchelaar NAD, Waling MM, Adhin AA, van Daele PLA, Schreurs MWJ, Rombach SM. The value of anti-neutrophil cytoplasmic antibodies (ANCA) testing for the diagnosis of ANCA-associated vasculitis, a systematic review and meta-analysis. Autoimmun Rev 20(1):102716, 2021. doi:10.1016/j.autrev.2020.102716
Treatment of Granulomatosis with Polyangiitis
Kidney transplantation if necessary
Treatment of granulomatosis with polyangiitis depends on the severity of disease. A multidisciplinary approach is required for multiorgan disease, often including a rheumatologist, otorhinolaryngologist, pulmonologist, and nephrologist.
Patients who have severe life-threatening or organ-threatening manifestationsRituximab, a B cell-depleting monoclonal antibody, is preferred because of the more favorable adverse effect profile compared with cyclophosphamide (1, 2). Efficacies of rituximab and cyclophosphamide appear to be similar for inducing remission (3). When cyclophosphamidecyclophosphamide4). Plasma exchange has not been shown to decrease the incidence of mortality or end-stage renal disease (5).
For maintenance therapy,rituximab is effective in lowering the risk of recurrence. In a randomized trial that included patients with GPA and other ANCA-associated vasculitides, patients treated with rituximab for maintenance therapy had fewer relapses and adverse events compared with those treated with azathioprine (6). The optimal dose, frequency of infusions, and duration of rituximab for maintenance therapy are not entirely clear. Patients with frequent relapses may need to take immunosuppressants indefinitely In one retrospective study, relapse rates were lower when rituximab was combined with methotrexate, azathioprine, or mycophenolate mofetil than when rituximab was used alone (7). A corticosteroid, given at a low dose, is often used to help maintain remission.
B cell-depleting therapies will markedly blunt the response to vaccines for months after administration and may cause hypogammaglobulinemia.
For less severe disease,Rituximab may be used instead of methotrexate. Corticosteroids are tapered to as low a dose as possible or discontinued.
Treatment of subglottic stenosis is difficult. Systemic immunosuppressants may not be effective. Intralesional injection of long-acting corticosteroids, with gentle progressive dilation, markedly improves outcome and limits the need for tracheostomy.
Patients should be taught about the disorder so that relapses can be detected early.
Kidney transplantation has been successful; the risk of relapse after transplantation is reduced compared with maintenance dialysis treatment (possibly due to use of immunosuppressants to prevent rejection).
Treatment references
1. Chung SA, Langford CA, Maz M, et al: 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol 73(8):1366-1383, 2021. doi:10.1002/art.41773
2. Kidney Disease: Improving Global Outcomes (KDIGO) ANCA Vasculitis Work Group. KDIGO 2024 Clinical Practice Guideline for the Management of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis [published correction appears in Kidney Int 2024 Jul;106(1):160-163. doi: 10.1016/j.kint.2024.04.003]. Kidney Int 2024;105(3S):S71-S116. doi:10.1016/j.kint.2023.10.008
3. Stone JH, Merkel PA, Spiera R, et al: Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 363:221–232, 2010. doi: 10.1056/NEJMoa0909905
4. Jayne DRW, Merkel PA, Schall TJ, Bekker P; ADVOCATE Study Group: Avacopan for the Treatment of ANCA-Associated Vasculitis [published correction appears in N Engl J Med 2024 Jan 25;390(4):388. doi: 10.1056/NEJMx230010]. N Engl J Med 384(7):599-609, 2021. doi:10.1056/NEJMoa2023386
5. Walsh M, Merkel PA, Peh CA, et al: Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med 382(7):622-631, 2020. doi:10.1056/NEJMoa1803537
6. Smith RM, Jones RB, Specks U, et al: Rituximab versus azathioprine for maintenance of remission for patients with ANCA-associated vasculitis and relapsing disease: an international randomised controlled trial. Ann Rheum Dis 82(7):937-944, 2023. doi:10.1136/ard-2022-223559
7. Sorin B, Samson M, Durel CA, et al: Rituximab plus methotrexate combination as a salvage therapy in persistently active granulomatosis with polyangiitis. Rheumatology (Oxford) 61(6):2619–2624, 2022. doi:10.1093/rheumatology/keab791
Prognosis for GPA
Prognosis depends on the severity and extent of disease and at least as much on how rapidly treatment occurs.
Use of immunosuppressants for severe disease has dramatically improved prognosis. With treatment, complete remission is possible for approximately 80% of patients, but approximately half of them eventually relapse (1); relapse may occur during remission maintenance therapy or after treatment is stopped (sometimes many years later). Resuming or increasing treatment can usually control the disorder. However, 90% of patients develop significant morbidity due to the disease and/or the treatments.
Prognosis reference
1. Alberici F, Smith RM, Jones RB, et al. Long-term follow-up of patients who received repeat-dose rituximab as maintenance therapy for ANCA-associated vasculitis. Rheumatology (Oxford) 2015;54(7):1153-1160. doi:10.1093/rheumatology/keu452
Key Points
In granulomatosis with polyangiitis, vasculitis affects small- and medium-sized vessels in any organ, typically the kidneys (with glomerulonephritis), and upper and lower respiratory tracts with significant necrotizing parenchymal granulomatous inflammation.
Manifestations can affect various organ systems and often include upper and lower respiratory tract symptoms (eg, recurrent nasal discharge or epistaxis, cough), followed by hypertension and edema (due to kidney involvement).
Confirm the diagnosis with antineutrophil cytoplasmic antibodies testing and biopsy.
Relapses are common, and treatments can contribute to morbidity.
Induce remission with corticosteroids plus another immunosuppressant.







