



Topic Resources


(See also Overview of Allergic and Atopic Disorders, Angioedema, and US HAEA [Hereditary Angioedema Association] Medical Advisory Board 2020 Guidelines for the Management of Hereditary Angioedema [2020].)
C1 inhibitor deficiency or dysfunction not only affects complement activation but also results in increased levels of bradykinin because C1 inhibitor inhibits activated kallikrein (required for the generation of bradykinin) in the kinin system pathway (1).
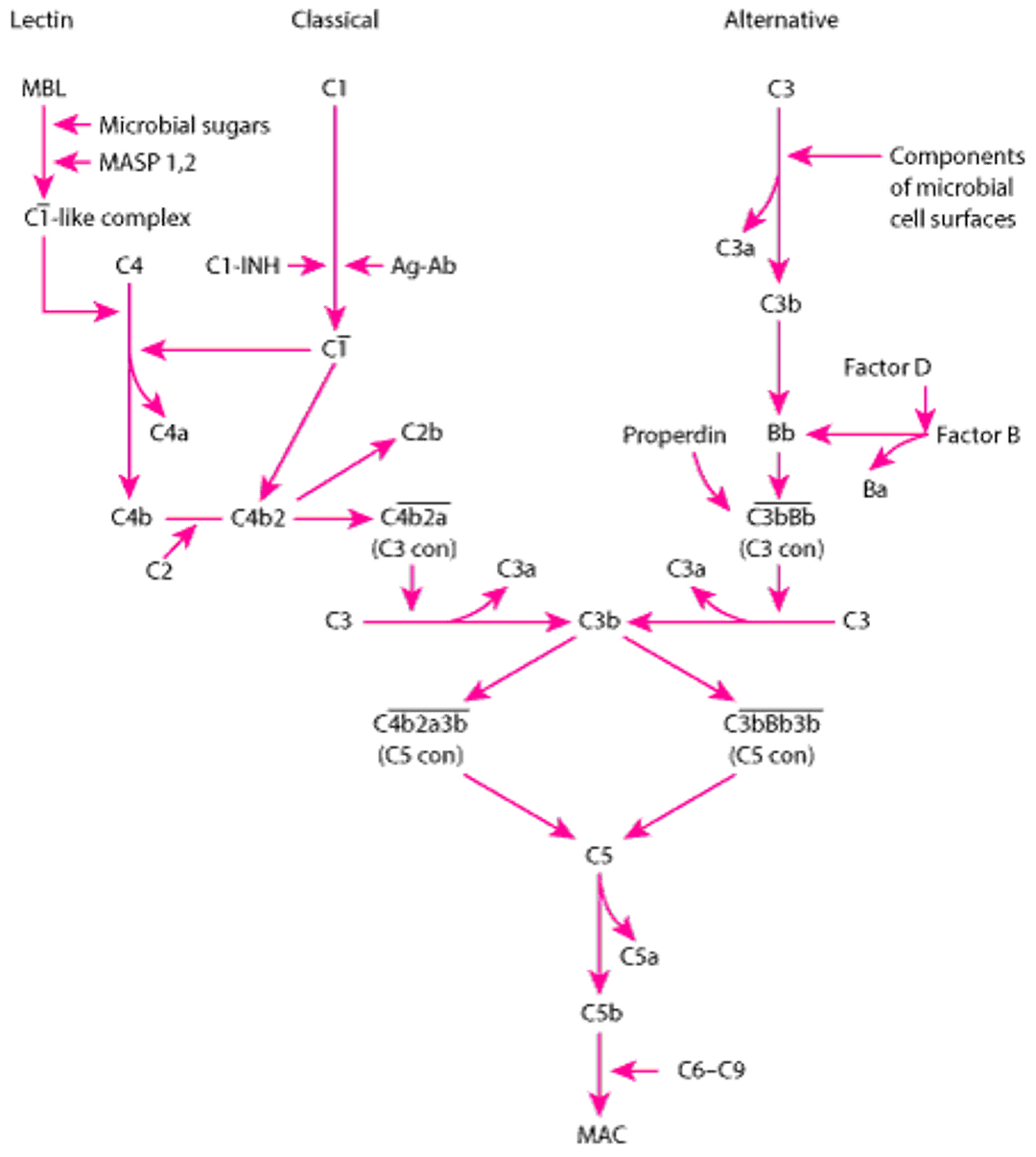
Complement Activation Pathways
The classical, lectin, and alternative pathways converge into a final common pathway when C3 convertase (C3 con) cleaves C3 into C3a and C3b. Ab = antibody; Ag =antigen; C1-INH =C1 inhibitor; MAC = membrane attack complex; MASP = MBL-associated serine protease; MBL = mannose-binding lectin. Overbar indicates activation. |

Hereditary angioedema
Hereditary angioedema has 3 types:
Type 1 (80 to 85%): Characterized by C1 inhibitor deficiency
Type 2 (15 to 20%): Characterized by C1 inhibitor dysfunction
Type 3 (rare): Characterized by normal C1 inhibitor function and levels
Type 1 and type 2 involve mutations of the gene encoding C1 inhibitor. Inheritance in type 1 is autosomal dominant. Clinical presentation is usually during childhood or adolescence; 75% of patients with type 1 have an episode by age 15 (1).
Type 2 results from a dysfunctional C1 inhibitor. Inheritance is autosomal dominant although de novo mutations occur in about 25% of cases (1).
Type 3 is rare. It is characterized by normal C1 inhibitor and is sometimes due to genetic mutations that result in abnormal forms of factor XII, plasminogen, angiopoietin 1, or kininogen. Type 3 occurs more frequently in females.
Acquired C1 inhibitor deficiency
C1 inhibitor deficiency may be acquired when
Complement is consumed in neoplastic disorders (eg, B-cell lymphoma, adenocarcinoma) or immune complex disorders.
C1 inhibitor autoantibody is produced in monoclonal gammopathy.
Rarely, C1 inhibitor autoantibody is produced in autoimmune disorders (eg, systemic lupus erythematosus [SLE], dermatomyositis).
Clinical presentation is usually at an older age, when patients have an associated disorder.
Triggers
In all forms of hereditary and acquired angioedema, attacks can be precipitated by
Mild trauma (eg, dental work, tongue piercing)
Viral illness
Cold exposure
Pregnancy
Estrogen-containing medications and tamoxifen -containing medications and tamoxifen
Ingestion of certain foods
Angioedema may be aggravated by emotional stress.
General reference
1. Miyata T, Horiuchi T. Biochemistry, molecular genetics, and clinical aspects of hereditary angioedema with and without C1 inhibitor deficiency. Allergol Int 2023;72(3):375-384. doi:10.1016/j.alit.2023.04.004
Symptoms and Signs of Hereditary and Acquired C1 Inhibitor DeficiencySymptoms and Signs of Hereditary and Acquired C1 Inhibitor Deficiency
Symptoms and signs of hereditary and acquired angioedema are similar to those of other forms of bradykinin-mediated angioedema, with asymmetric and mildly painful swelling that often involves the face, lips, and/or tongue. Swelling may also occur on the back of hands or feet or on the genitals.
The gastrointestinal tract is often involved, with variable manifestations that suggest intestinal obstruction, including nausea, vomiting, and colicky discomfort.
Pruritus, urticaria, and bronchospasm do not occur, but laryngeal edema may be present, causing stridor (and sometimes death).

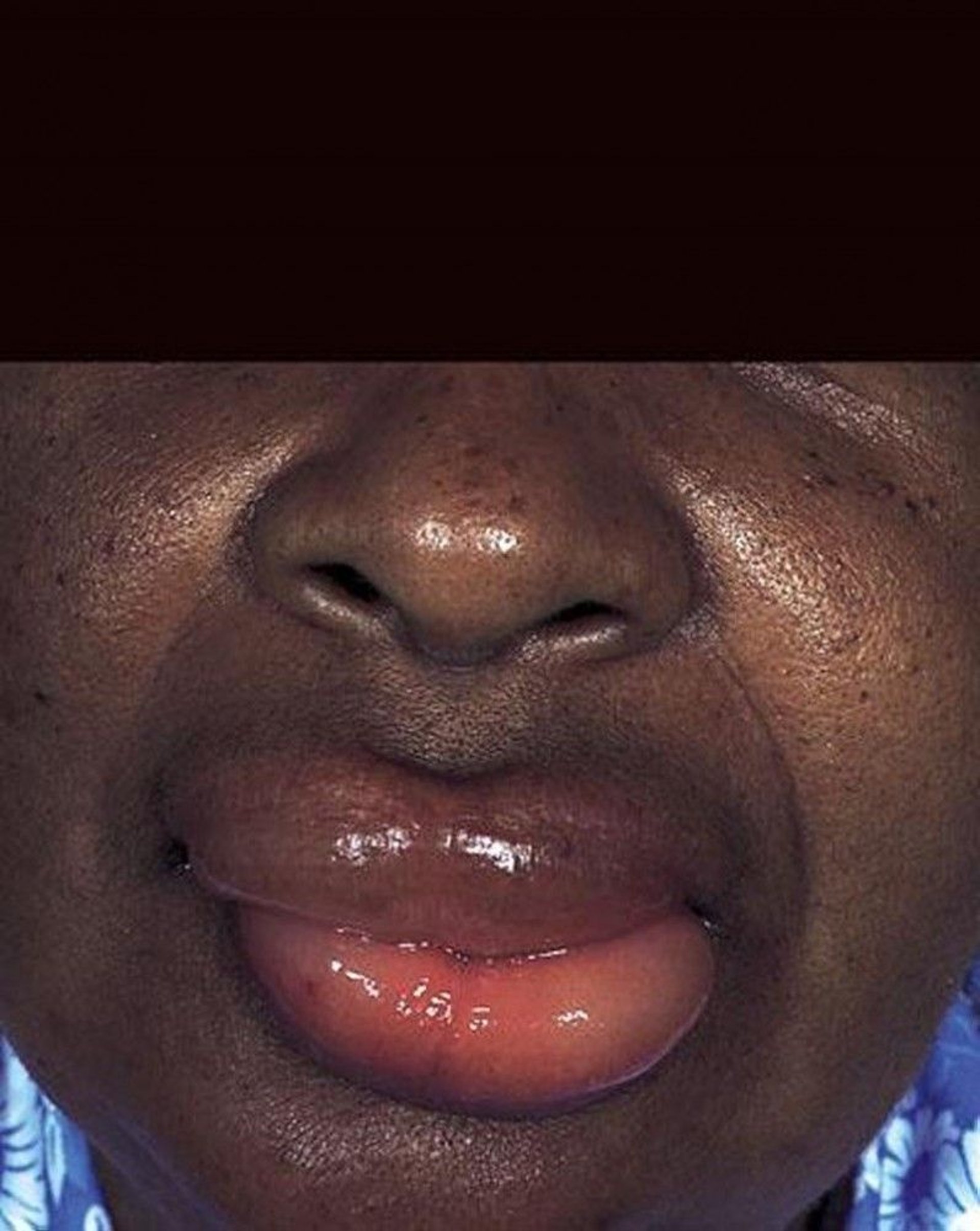
By permission of the publisher. From Joe E, Soter N. In Current Dermatologic Diagnosis and Treatment, edited by I Freedberg, IM Freedberg, and MR Sanchez. Philadelphia, Current Medicine, 2001.
Swelling resolves within about 1 to 3 days of onset. In hereditary angioedema, symptoms resolve as complement components are consumed.
Diagnosis of Hereditary and Acquired C1 Inhibitor DeficiencyDiagnosis of Hereditary and Acquired C1 Inhibitor Deficiency
Measurement of complement levels
If angioedema is not accompanied by urticaria and recurs without any clear cause or is triggered by local trauma, clinicians should suspect hereditary or acquired C1 inhibitor deficiency.
Levels of C4, C1 inhibitor, and C1q (a component of C1) are measured. Hereditary angioedema (types 1 and 2) or acquired C1 inhibitor deficiency is confirmed by
Low levels of C4, even between episodes
Decreased C1 inhibitor level or function
Other findings include
Type 1 hereditary C1 inhibitor deficiency: Low C1 inhibitor protein levels, decreased C1 inhibitor function, and normal C1q levels
Type 2 hereditary C1 inhibitor deficiency: Normal or increased C1 inhibitor protein levels, decreased C1 inhibitor function, and normal C1q levels
Acquired C1 inhibitor deficiency: Low C1q levels
Type 3 hereditary C1 inhibitor deficiency: Normal C1 inhibitor level, C1 inhibitor function, and C1q levels
All first-degree relatives of patients with confirmed hereditary C1 inhibitor deficiency should be screened whether they have symptoms or not. Screening should include C1 inhibitor and C4 levels (1).
Diagnosis reference
1. Zuraw BL, Bernstein JA, Lang DM, et al: A focused parameter update: Hereditary angioedema, acquired C1 inhibitor deficiency, and angiotensin-converting enzyme inhibitor–associated angioedema. J Allergy Clin Immunol 131 (6):1491-1493, 2013. doi: 10.1016/j.jaci.2013.03.034
Treatment of Hereditary and Acquired C1 Inhibitor DeficiencyTreatment of Hereditary and Acquired C1 Inhibitor Deficiency
For acute attacks, C1 inhibitor, ecallantide, icatibantFor acute attacks, C1 inhibitor, ecallantide, icatibant
For acute attacks, the following are considered first-line treatment:
Purified plasma-derived human C1 inhibitorPurified plasma-derived human C1 inhibitor
Recombinant C1 inhibitor obtained from the milk of transgenic rabbitsRecombinant C1 inhibitor obtained from the milk of transgenic rabbits
C1 inhibitor obtained from human plasma C1 inhibitor obtained from human plasma
Ecallantide (a recombinant protein that acts as a reversible inhibitor of kallikrein)Ecallantide (a recombinant protein that acts as a reversible inhibitor of kallikrein)
Icatibant (a synthetic decapeptide that acts as a reversible competitive antagonist of the bradykinin type 2 receptor)Icatibant (a synthetic decapeptide that acts as a reversible competitive antagonist of the bradykinin type 2 receptor)
Recombinant C1 inhibitor has similar protease inhibitory activity but a shorter half-life than plasma-derived C1 inhibitor (Recombinant C1 inhibitor has similar protease inhibitory activity but a shorter half-life than plasma-derived C1 inhibitor (1).
If none of these medications is available, fresh frozen plasma or, in the European Union, tranexamic acid has been used. If none of these medications is available, fresh frozen plasma or, in the European Union, tranexamic acid has been used.
If the airways are affected, securing an airway is the highest priority. Epinephrine may provide transient benefit in acute attacks when airways are involved. However, the benefit may not be sufficient or may be temporary; then endotracheal intubation may be necessary. Corticosteroids and antihistamines are not effective.If the airways are affected, securing an airway is the highest priority. Epinephrine may provide transient benefit in acute attacks when airways are involved. However, the benefit may not be sufficient or may be temporary; then endotracheal intubation may be necessary. Corticosteroids and antihistamines are not effective.
Analgesics, antiemetics, and fluid replacement can be used to relieve symptoms.
Treatment of patients with hereditary C1 inhibitor deficiency focuses on 4 core principles (2):
Availability of effective on-demand acute therapy for all patients
Early treatment to prevent attack progression
Treatment of attacks regardless of the site of swelling
Incorporation of long-term prophylaxis based on highly individualized decision-making reflecting a physician-patient partnership
Based on these principles, all patients with confirmed hereditary angioedema should have access to at least 2 standard doses of an on-demand medication for treatment of acute attacks (2).
Pearls & Pitfalls
|
Treatment references
1. Moldovan D, Bernstein JA, Cicardi M: Recombinant replacement therapy for hereditary angioedema due to C1 inhibitor deficiency. Immunotherapy 7 (7):739–752, 2015. doi: 10.2217/imt.15.44
2. Busse PJ, Christiansen SC, Riedl MA, et al: US HAEA Medical Advisory Board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract 9 (1):132–150.e3, 2021. doi: 10.1016/j.jaip.2020.08.046
Prevention of Hereditary and Acquired C1 Inhibitor DeficiencyPrevention of Hereditary and Acquired C1 Inhibitor Deficiency
Long-term prophylaxis
Medications used for long-term prophylaxis of hereditary C1 inhibitor deficiency episodes include
Plasma-derived C1 inhibitor (human)Plasma-derived C1 inhibitor (human)
LanadelumabLanadelumab
BerotralstatBerotralstat
Attenuated androgens
Antifibrinolytics (eg, tranexamic acid)Antifibrinolytics (eg, tranexamic acid)
Plasma-derived C1 inhibitor may be given in regular IV infusions or subcutaneous injections as long-term prophylaxis. Patients can be taught to self-administer. Plasma-derived C1 inhibitor is available for long-term prevention of hereditary angioedema in the United States, but recombinant C1 esterase inhibitor is not.Plasma-derived C1 inhibitor may be given in regular IV infusions or subcutaneous injections as long-term prophylaxis. Patients can be taught to self-administer. Plasma-derived C1 inhibitor is available for long-term prevention of hereditary angioedema in the United States, but recombinant C1 esterase inhibitor is not.
Lanadelumab is a recombinant humanized monoclonal antibody that binds to plasma kallikrein and blocks its activity.Lanadelumab is a recombinant humanized monoclonal antibody that binds to plasma kallikrein and blocks its activity.
Berotralstat is a synthetic small molecule developed to inhibit plasma kallikrein.Berotralstat is a synthetic small molecule developed to inhibit plasma kallikrein.
Attenuated androgens (eg, stanozolol, danazol) are used to stimulate hepatic Attenuated androgens (eg, stanozolol, danazol) are used to stimulate hepaticC1 inhibitor synthesis. This treatment may be less effective for the acquired form of angioedema.
Antifibrinolytics (eg, tranexamic acid) have been used as second-line medications for long-term prophylaxis in children and patients who are pregnant.Antifibrinolytics (eg, tranexamic acid) have been used as second-line medications for long-term prophylaxis in children and patients who are pregnant.
Short-term prophylaxis
Short-term prophylaxis for hereditary C1 inhibitor deficiency is indicated before high-risk procedures (eg, dental or airway procedures) if C1 inhibitor is not available to treat an acute attack. Patients are usually given attenuated androgens (eg, danazol, stanozolol) 5 days before the procedure until 2 days afterward. If C1 inhibitor (plasma-derived or recombinant) is available, some experts advocate giving it 1 hour before high-risk procedures rather than attenuated androgens for short-term prophylaxis. Plasma products (eg, 2 units of fresh frozen plasma) before procedures are also an option (deficiency is indicated before high-risk procedures (eg, dental or airway procedures) if C1 inhibitor is not available to treat an acute attack. Patients are usually given attenuated androgens (eg, danazol, stanozolol) 5 days before the procedure until 2 days afterward. If C1 inhibitor (plasma-derived or recombinant) is available, some experts advocate giving it 1 hour before high-risk procedures rather than attenuated androgens for short-term prophylaxis. Plasma products (eg, 2 units of fresh frozen plasma) before procedures are also an option (1).
Prevention reference
1. Prematta M, Gibbs JG, Pratt EL: Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 98 (4):383–388, 2007.
Key Points
Onset is usually during childhood or adolescence for hereditary angioedema or during later adulthood for acquired angioedema, often in patients with a neoplastic or an autoimmune disorder.
Mild trauma, viral illness, cold exposure, pregnancy, or ingestion of certain foods may trigger attacks; emotional stress may aggravate them.
Measure complement levels; low levels of C4 and decreased C1 inhibitor function indicate hereditary angioedema or acquired C1 inhibitor deficiency.
For acute attacks, use purified human C1 inhibitor, recombinant C1 inhibitor, ecallantide, or icatibant, and for symptom relief, use analgesics, antiemetics, and fluids; antihistamines and corticosteroids are ineffective. For acute attacks, use purified human C1 inhibitor, recombinant C1 inhibitor, ecallantide, or icatibant, and for symptom relief, use analgesics, antiemetics, and fluids; antihistamines and corticosteroids are ineffective.
For long-term prophylaxis, use regular infusions of plasma-derived C1 inhibitor, lanadelumab, or berotralstat.For long-term prophylaxis, use regular infusions of plasma-derived C1 inhibitor, lanadelumab, or berotralstat.
For short-term prophylaxis (eg, before dental or airway procedures), consider C1 inhibitor, attenuated androgens (eg, stanozolol, danazol), or plasma products such as fresh frozen plasma.For short-term prophylaxis (eg, before dental or airway procedures), consider C1 inhibitor, attenuated androgens (eg, stanozolol, danazol), or plasma products such as fresh frozen plasma.






